
Abstract
In this work, a highly sensitive solid-phase-dispersive microextraction method was designed based on magnetic carbon nanocomposites as a magnetic solid-phase extraction sorbent coupled with dispersive liquid–liquid microextraction and two miscible stripping solvents (MSPE–DLLME) followed by gas chromatography–mass spectrometry (GC–MS) for determination of 16 polycyclic aromatic hydrocarbons (PAHs). By adopting this research methodology, a mixture of two miscible organic solvents is used not only as stripping solvent for MSPE, but also as extraction and disperser solvents for DLLME procedure. Several parameters such as amount of extraction adsorbent, type of stripping, extraction solvents and their volumes, salt effect, and pH and volume of sample solution were optimized to obtain high extraction recoveries. Finally, 2 µL of extraction phase was injected into GC–MS. Under optimal conditions, the method attained satisfactory precisions (RSD% ≤ 8.66), excellent limits of detection in the range of 0.1–0.5 ng kg−1 at S/N = 3, and very high enrichment factors in the range of 28,187–33,149 for 500 mL sample solution of different PAHs. The calibration curves of 16 extracted PAHs were linear in the range of 0.4–10,000 ng kg−1, with coefficients of determination (r2) between 0.9989 and 0.9999. The optimized method to determine 16 PAHs has been successfully applied in the real environment including waters, waste water, sewage, and soil.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are hazardous compounds; carcinogenesis has been reported for tri-, tetra-, penta-, and hexa-cyclic compounds [1]. PAHs consisting of two or more fused aromatic rings are present everywhere in the environment. These compounds are produced by incomplete combustion of organic matters and after concentration by biological chain, and they possess high mutagenicity and carcinogenicity effects. Because of their destructive effects such as oxidative stress and oxidative DNA damage through metabolic activation and the generation of reactive oxygen species, special attention has been paid to the monitoring of PAHs as environmental pollutants.
The Environmental Protection Agency (EPA), 40 CFR Part 136, guidelines establishing test procedures for the analysis of pollutants under the clean water act has promulgated 16 PAHs as very important contaminants [2, 3]. Due to very low concentration levels and tracking their wide distribution in complex environmental samples, the development of pre-concentration methods and clean-up procedures combined with very selective and sensitive analysis techniques are necessary. Common methods for the extraction and concentration of PAHs and similar compounds from real environmental samples are as follows: liquid–liquid extraction (LLE) [4], solid-phase extraction (SPE) [1, 2, 5, 6], headspace solvent microextraction (HSME) [7], magnetic solid-phase extraction (MSPE) [8,9,10,11], homogeneous liquid–liquid microextraction [12], dispersive liquid–liquid microextraction (DLLME) [13,14,15], liquid-phase microextraction methods based on solidification of floating organic drop (SDME) [16], ultrasound-assisted emulsification microextraction (USAEME) [17], stirring bar sorptive extraction (SBSE) [18, 19], solid-phase microextraction (SPME) [20], headspace SPME [21], hollow fiber liquid-phase microextraction (HF-LPME) [22], headspace sorptive extraction [23], and coupling methods such as solid-phase extraction combined with DLLME [24] and magnetic solid-phase extraction coupled with DLLME [25].
Analysis of PAHs in various sample matrices is feasible after pre-concentration stage using high-performance liquid chromatography–fluorescence (HPLC–FLD) [9, 26], ultraviolet (HPLC–UV) [6] or diode-array detectors (HPLC–DAD) [10], gas chromatography–mass spectrometry (GC–MS) [13, 23, 27] or (GC–MS/MS) [28], gas chromatography–flame ionization detector (GC–FID) [25], GC and HPLC coupled to electrospray ionization mass spectrometry (HPLC–ESI–MS), and ultra-performance liquid chromatography–diode-array detector (UPLC–DAD) [10].
Two of the most efficient methods for separation of environmental pollutants from water are MSPE and DLLME. The separation of trace analytes from large volumes of solution can take a lot of time using standard SPE column. However, in MSPE procedure, magnetic adsorbents with a large specific surface area added to the solution. The analytes are adsorbed on to the magnetic adsorbent and then the adsorbent with adsorbed analytes is recovered from the suspension using an appropriate magnetic separator easily. The analytes are eluted from the adsorbent with an extracting solvent. DLLME is a microextraction technique using microliter volumes of the extraction solvent. This method includes a binary mixture of the extraction and disperser solvents, which are dispersed in aqueous media. The analytes are extracted into the fine droplets of the extraction solvent in microliter amounts.
For the first time in this study, a combined pretreatment technique of MSPE and DLLME based on the use of magnetic carbon nanocomposites and two miscible organic stripping solvents (MSPE–DLLME) was designed as the ultra–pre-concentration method for 16 PAHs followed by gas chromatography–mass spectrometry (GC–MS). In this methodology, two miscible organic solvents act not only as stripping solvent for MSPE, but also as extraction and disperser solvents for DLLME procedure. The parameters affecting the efficiency of the extraction method were studied and optimized. Finally, optimized method used to the highly sensitive determination of PAHs in tap water, sea water, waste water, sewage, and soil samples.
Experimental
Chemicals and Instrumentation
Activated carbon and ferric nitrate Fe(NO3)3·9H2O for the synthesis of magnetic carbon nanocomposites (MCNs) were prepared from Merck (Darmstadt, Germany). Calibration mixture of 16 PAHs’ standards dissolved in acetonitrile and biphenyl (as internal standard) were purchased from Supelco (Bellefonte, PA, USA). These PAHs, include naphthalene (Nap), acenaphthylene (Acy), acenaphthene (Ace), fluorene (Fl), phenanthrene (Phe), anthracene (Ant), fluoranthene (Flu), pyrene (Py), benz(a)anthracene (BaA), chrysene (Chr), benzo(b)fluoranthene (BbF), benzo(k)fluoranthene (BkF), benzo(a)pyrene (BaP), indeno(1,2,3–cd) pyrene (IPy), dibenz(a,h)anthracene (DahA), and benzo(ghi)perylene (BghiP). The concentration of each PAH in the mixture was 2000 mg L−1. Stock solution of 10 mg L−1 was prepared with HPLC grade acetonitrile, and working standard solutions were prepared daily with doubly distilled water. Toluene, benzene, n-hexane, ethyl acetate, dichloromethane, acetonitrile, ethanol, methanol, acetone, and other solvents were purchased from Merck (Darmstadt, Germany). All other chemical materials used the highest purity too.
The laboratory apparatus used included a 35 kHz ultrasonic water bath with temperature control (Bandelin Sonorex Digital, Germany), a digital IKA vortex (MS3 basic), and an IKA shaker (Deutschland, Germany) for stirring the sample solutions and a centrifuge from Parsia Ind. Group (Tehran, Iran) to separate the extractant from the sample solution. A Nd–Fe–B strong magnet (10 cm × 5 cm × 4 cm, 1.47 T) was used for separation of sorbent from water samples. The pH of solutions was adjusted by a Metrohm 781 ion analyzer (Herisau Switzerland) with a combined glass-calomel electrode. Twelve milliliters home-made glass vials were used for extraction procedures.
An FT-IR Spectrometer Bruker VERTEX 70, equipped with a Globar source, a DGTS detector, was used to record the IR spectra in the range of 500–4000 cm−1.
GC–MS Analysis
Separation, determination, and identification of the target analytes were performed using an Agilent 7890A gas chromatograph which was equipped with a 5975 mass selective detector (MSD, Agilent Technologies) and fitted with Restek Rxi®-5MS fused-silica capillary column 5% Phenyl Methyl Silox (60 m × 0.32 mm i.d. and 0.25 µm film thickness). Carrier gas, helium (purity 99.999%), was set at a flow rate of 1.0 mL min−1. The injector operated in the splitless mode with an injector temperature of 290 °C. The temperature of oven set at 70 °C (for 2 min), following an increased ramp of 20 °C min−1 to 220 °C, and held for 2 min, then to 295 °C at a rate of 5 °C min−1 for 8 min, and the solvent delayed for 4 min, for a total run time of 34.5 min. The MSD operated in electron impact (EI) mode at 70 eV with an ion source temperature of 230 °C. MSD transfer line and quadruple temperatures were 290 and 150 °C, respectively. The samples and standard solutions were injected (2 µL) into the GC–MS, after extraction. For quantitative analysis, the MSD system was configured in selective ion monitoring (SIM) mode. The target ion determined by injection of PAHs standard under the same program but in the scan conditions from 40 to 550 m/z.
Preparation and Characterization of Magnetic Carbon Nanocomposite
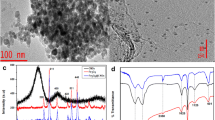
In this work, activated carbon-based magnetic nanocomposite with exploitable characteristics was prepared and used as adsorbent for pre-concentration of PAHs from aqueous samples. These magnetic carbon nanocomposites’ (MCNs) adsorbent include commercially activated carbon and available magnetic iron oxide nanoparticles. In addition, they were carefully characterized before checking their adsorption properties for the extraction and pre-concentration of these compounds. The magnetic carbon nanocomposites were prepared by the method already explained by Tajabadi et al. [29]. Initially, activated carbon dispersed in concentrated nitric acid (65%) for 3–4 h and heated at 80 °C to make it hydrophilic. Then, 1 g of the modified carbon was added to 40 mL solution containing 8 g Fe(NO3)3·9H2O and sonicated for 45 min to adsorb the iron salt. Then, the resulted mixture was filtrated dried, and then modified activated carbon heated at 750 °C for 3 h under N2 atmosphere. Figure 1a illustrates the existence of Fe3O4 nanoparticles. The magnetic hysteresis loop of MCNs is checked out by a vibrating sample magnetometer (VSM) shown in Fig. 1b. The report of magnetization (M) vs magnetic field (H) shows very weak hysteresis by revealing that the resultant magnetic nanoparticles are approximately superparamagnetic with a saturation magnetization (Ms) of 10.5 emu g−1 at room temperature. The presence of peaks at 585.78–636.95 wavenumber cm−1 FT-IR spectrum of the MCNs compared to CNs (Fig. 1c) well indicated that there are iron oxides inside the synthesized nanoparticles [11, 29].

a TEM image of the prepared MCNs, b VSM diagram of MCNs, and c FT-IR spectra of the CNs (yellow) and MCNs (violet)
Pretreatment of Real Samples
The original water samples collected were stored in pre-cleaned polyethylene bottles in a fridge at about 4 °C under darkness condition. For preparation of soil samples, first, the sample was air dried and homogenized. 30 g of sample was weighed in a 200-mL beaker, and 50 mL of a proper n-hexane/dichloromethane mixture was added. Then, the sample was ultrasonically extracted for 3 min. The extract was decanted into a centrifuge bottle and centrifuged to remove particles. The extraction was repeated two or more times with two additional 50 mL portions of the solvent. After each ultrasonic extraction, the solvent was decanted. After the final ultrasonic extraction, the sample was poured into the Buchner funnel and rinsed with extraction solvent. A vacuum was then applied to the filtration flask, and the solvent extract was collected. Finally, the extracting solvent evaporated by a stream of nitrogen gas and remaining analyte dissolves in approximately 2 mL acetone. This extract sample was poured into a 500 mL flask and diluted to the mark with HPLC grade water.
MSPE–DLLME Procedure
In the present study, efficient magnetic carbon nanocomposites used as sorbent for simple, rapid, and effective microextraction of PAHs from environmental samples. To achieve high extraction recoveries, the target analytes on the adsorbent were eluted by two miscible stripping solvents, and then, extraction solvent was separated in a liquid–liquid dispersion process. The presence of toluene in acetonitrile as two miscible stripping solvents led to the best elution of PAHs. An aliquot of 500 mL of aqueous sample transferred into a proper Erlenmeyer flask. Then, 25 mg of magnetic carbon nanocomposite sorbent added into it and the resulted mixture was completely shaken for 30 min. After that, a Nd–Fe–B strong magnet (10 cm × 5 cm × 4 cm, 1.47 T) was put at the bottom of the Erlenmeyer flask, and the magnetic carbon nanocomposites were collected from the solution. After about 3 min, the solution became smooth and the top of clear solution was decanted. Then, the adsorbed target analytes were eluted from the sorbent by two miscible stripping solvents, including 1.5 mL acetonitrile containing 50 µL toluene and 1.0 mg L−1 biphenyl, as an internal standard, by 15 min sonication at 70 °C. Next, the solvent mixture suddenly injected into 12 mL doubly distilled water. In the test tube, a cloudy solution was formed and the mixtures were centrifuged for 8 min at 4000 rpm. Accordingly, the extraction solvent droplet placed on the surface of the solution because of its low density. Finally about 15 µL from extraction solvent was raised into the capillary tube of the glass vial, and then, this extraction phase was collected by a 25-µL syringe and 2 µL from it was injected into GC–MS instrument with a PAL system (CTC analytics, AG, Switzerland) as an auto injector system.
Results and Discussion
Optimization of MSPE–DLLME Parameters
In addition to the MSPE parameters such as type and volume of solvents that contained stripping and extraction solvents for DLLME, the pH of initial sample solution, extraction time, amount of magnetic sorbent and break through volume, elution condition, salt effect, and time of centrifugation in liquid–liquid dispersion process were also investigated and optimized in aqueous samples. All optimization experiments were performed on GC–MS.
Effect of Stripping and Extraction Solvents
In this work, two miscible stripping solvents (disperser and extraction solvents used in DLLME process) were used for elution of PAHs from magnetic carbon nanocomposites, and then, the extraction solvent containing concentrated target analytes was separated from disperser solvent (acetonitrile) by DLLME method. Both of these solvents should be able for elution, but the main elution solvent is the extraction solvent for DLLME in microliter volume and other solvent was used to increase the contact area. The disperser part of stripping solvents in the first stage of the extraction procedure (MSPE) plays two roles, the main role of disperser part is extraction solvent dispersion between particles of magnetic carbon nanocomposites to increase contact on the one hand, and also achieving higher solubility in water, so that it can be separated from extraction solvent by liquid–liquid dispersion process on the other hand. Finally, it should have extraction ability of the PAHs (in MSPE) and be miscible with organic solvent, so that it plays the role of disperser solvent in DLLME process, as well. For this purpose, methanol, ethanol, acetone, and acetonitrile were investigated. Acetonitrile indicated the highest extraction efficiency, and thus, it was selected as the disperser part of stripping solvent for the subsequent experiments due to its good elution ability and forming a proper cloudy solution in liquid–liquid dispersive step. Figure 2 shows the peak area of the extracted analytes for each solvent.

Effects of type of disperser solvent on the extraction efficiency of PAHs. Conditions: for extraction; disperser solvent volume, 1.5 mL; organic extraction solvent, 50 µL; amount of MCNs, 25 mg; sample solution volume, 500 mL; for elution; sonication time 15 min at 70 °C; centrifuge time, 8 min at 4000 rpm
The use of acetonitrile (ACN) compared to other organic solvents as striping solvent not only improved the extraction efficiency but also provided the need for a lower volume of organic solvents. Suitable extraction solvent is also very important, it must possess low water solubility, the ability for elution of the PAHs from sorbent, and also compatibility with the analytical instruments to be used. Due to the structure of PAHs, different types of extraction solvents, lighter and heavier than water, have been reported for use in the DLLME procedure [17, 19, 30, 31]. In this work, among the most common organic solvents, a number of solvents with higher and lower density from water were examined on the basis of the extraction capability of the analytes of interest and also proper gas chromatography behavior. Thus, the next step investigated the behavior of the mixture of acetonitrile with some miscible organic solvents including tetrachloroethylene (density 1.62 g mL−1), chlorobenzene (density 1.11 g mL−1), 1-octanol (density 0.82 g mL−1), n-hexane (density 0.659 g mL−1), and toluene (density 0.86 g mL−1) as stripping solvents. As can be seen in Fig. 3, the mixture of acetonitrile with toluene revealed the best extraction efficiency. Therefore, this mixture was selected as stripping solvents, and also disperser and extraction solvents for the next DLLME step.

Effects of type of stripping solvent on the extraction efficiency of PAHs. Conditions: for extraction; disperser solvent volume, 1.5 mL; organic extraction solvent, 50 µL; amount of MCNs, 25 mg; sample solution volume, 500 mL; for elution; sonication time 15 min at 70 °C; centrifuge time, 8 min at 4000 rpm
Effect of Volume of Stripping Solvents
To optimize the volume of extraction solvent, other experiments were performed using 1.5 mL ACN (stripping solvent) and different volumes of toluene (30.0, 40.0, 50.0, 60.0, and 70.0 µL). The results exhibited a dramatic increase in extraction efficiencies with the increasing volume of the extraction solvent to 50 µL. Due to the dilution effect, however, the peak area of PAHs in the extraction solvent was reduced by increasing the volume of the organic phase. To achieve more reproducible and easier collection, a volume of 50 µL of toluene was chosen as the optimum volume. For optimized volume of disperser solvent, other experiments were performed using various volumes of ACN (0.5, 1.0, 1.5, and 2.0 mL). Finally, based on the results obtained, 1.5 mL of ACN was selected as volume of disperser solvent.
Effect of the Desorption Conditions
For this purpose, the sorbent was eluted by a mixture of 1.5 mL ACN and 50 µL toluene by two methods (i.e., vortex and sonication at 10 min), in which the eluting of sorbent by sonication was found to be superior over vortex method. In the next step, the time and temperature of sonication were optimized. Temperature was expected to have a measurable effect on the distribution coefficient, desorption factor, and the mass transfer of the components. To optimize the temperature of sample solution during the elution process, the ultrasonic water bath was switched on at maximum frequency and power at various temperatures from 30 to 80 °C. The results showed the best extraction efficiency at 70 °C. The desorption time was also tested from 5 to 20 min. Finally, the results clearly showed that the best elution of sorbents by the binary solvent mixture occurred in 15 min sonication at 70 °C producing better extraction efficiencies.
Effect of Sample Solution pH
The pH of sample solution was adjusted from 2.0 to 10.0, to investigate the effect of pH on the extraction recoveries. However, no significant change was observed in the results. Thus, there was no need to pH control in the future experiments.
Effect of the Breakthrough Volume
To analyze the ultra-traces of pollutant materials and to get satisfactory recoveries for analytes, the use of high volume sample solution is necessary; since the MCNs adsorbents are separated by magnets, it was possible to collect the sorbent from larger volumes of the sample solution. Five different volumes (200, 300, 400, 500, and 600 mL) of aqueous samples were investigated three times for the breakthrough volume, each one being spiked with 200 ng of individual PAHs. It was found that the best pre-concentration factors can be obtained when the sample volumes were 500 mL (see Fig. 4). Thus, this volume was selected for further studies.

Effect of sample volume on the extraction efficiency of PAHs. Conditions: 200 ng of individual PAHs in each sample volume; disperser solvent (ACN) volume, 1.5 mL; organic extraction solvent (toluene) volume, 50 µL; amount of MCNs, 25 mg; elution condition, 15 min sonication at 70 °C; 8 min centrifuge at 4000 rpm
Effect of Amount of Adsorbent
Different amounts of magnetic carbon nanocomposite (MCNs) from 10 to 30 mg were added to 500 mL solution of PAHs (2 µg L−1). As a result of increase in surface of sorbent for adsorbing analytes, the recoveries were found to improve by increasing the amount of the adsorbent from 10 to 25 mg, due to expected increasing extraction recoveries. However, a further increase in amount of sorbent amount found to decrease the recoveries because of insufficient elution of sorbent with the stripping solvents. Therefore, 25 mg was chosen as the optimized mass of the sorbent for further studies.
Effect of Extraction Time in MSPE
To determine the effect of extraction time on the PAHs’ adsorption from 500 mL sample solution, different contact time (10, 15, 20, 25, 30, and 35 min) were examined. The recoveries reached to maximum at 30 min. Due to the high volume and low mass absorbent, the choice of this time is inevitable. Then, 30 min was selected as the optimal time.
Salt Effect in DLLME
In this work, the aim of controlling ionic strength is a better insulation of extraction solvent and preventing the analytes in aqueous phase. The extraction efficiency of PAHs was not changed by salt addition; this effect was evaluated by increasing NaCl concentrations in the range of 0–30% (w/v) in sample solution. On the other hand, increasing the salt concentration decreased the amount of collected solvent. Therefore, this extraction procedure was carried out without any addition of salt.
Effect of Extraction Time and Centrifugation Speed in DLLME
Final step of the MSPE–DLLME procedure shows the extraction solvent dispersion in the aqueous phase and the formation of the cloudy phase. In this sense, at highest speed of centrifuge (4000 rpm), the efficiency of method was examined at different times. However, the extraction time did not affect the performance of the method, and, therefore, a practical time of 8 min was selected.
Analytical Performance of MSPE–DLLME
The analytical characteristics of the presented method, i.e., linear dynamic range (LDR) limits of detection (LOD), limits of quantification (LOQ), pre-concentration factor (PF), and relative standard deviation (RSD) for all PAHs studied were obtained in aqueous samples under optimized conditions, and the results have been summarized in Table 1. This table also includes the corresponding quantification and identification ions (m/z values) of the 16 PAHs. To evaluate the linearity of the method, 10 spiking level of 16 PAHs in the concentration range of 0.1–10,000 ng kg−1 were used and LDRs in the range of 0.4–10,000 ng kg−1 were obtained for water samples. The LODs for all PAHs were practically evaluated based on a signal-to-noise ratio of 3, being in the range of 0.1–0.3 ng kg−1. At 400 and 2000 ng kg−1 levels, the intra-day and inter-day precisions were investigated by five replicate extractions. All RSDs% obtained were in the satisfactory range of 0.46–9.07%. The pre-concentration factors (PFs) which calculated based on Eq. 1 were found to be quite high, between 28, 187 and 33, 149:
where Cex,final is the final concentration of the analyte in the extractionsolution, and Cd,initial is the initial analyte concentration in the donor phase.
Comparison of MSPE–DLLME with Other Methods
The essential analytical characteristics of the method were compared with the previous techniques for determination of PAHs in environmental samples, based on solid-phase microextraction [20], head space solvent microextraction [7], dispersive liquid–liquid microextraction [30], and other methods [10, 17, 25, 29]; the results are shown in Table 2. As is obvious, RSDs are better or comparable with those of other studies, while the PFs reported here are much larger than the previously reported ones. Then, this MSPE–DLLME/GC–MS is a sensitive method for the pre-concentration and determination of PAHs from environmental water and soil samples. Furthermore, the number and variety of investigated real samples in this study are more than those in the previous works.
Analysis of Real Samples
Analysis of Environmental Water Samples
To verify the performance of the method, four aqueous samples including tap water sample from Karaj (Iran), industrial waste water from Tehran (Iran), sea water from Anzali (Iran), and mud river water from Karoon river, Ahvaz (Iran) were collected and analyzed through the procedure described in “Pretreatment of real samples”. The results showed that tap water was free from PAHs, but some of PAHs were detected in other water samples (see Table 3). To check the method recovery, the water samples were spiked with the PAHs contaminant at two various concentrations (i.e., 80 and 400 ng kg−1). The relative recoveries (RR%) and relative standard deviation (RSD%) of analytes were in the range of 74.8–102.6 and 0.7–12.8%, as shown in Table 3. It is obvious that this method is applicable to any type (salty and sweet) of water and waste water samples. Figure 5a shows the chromatograms obtained for industrial waste water blank and spiked of 400 ng kg−1 for each of PAHs.

Chromatograms of waste water and spiked waste water, with 400 ng kg−1 of each PAH obtained by MSPE–DLLME/GC–MS method. Extraction conditions: extraction solvent (toluene) volume, 50 µL; disperser solvent (ACN) volume, 1.5 mL; sample solution volume, 500 mL; amount of MCNs, 25 mg; elution condition, 15 min sonication at 70 °C; 8 min centrifuge at 4000 rpm
Analysis of Soil Samples
The extraction protocol is optimized for liquid samples, to evaluate the efficiency of the method for soil samples, it is necessary that the soil sample is first prepared and target analytes is transferred to the aqueous solution. The analytical characteristics listed in Table 1 are not applicable to non-aqueous samples. A soil sample collected from Oil Region-Ahvaz (Iran) and pretreatment was performed according to USEPA Method 3550B (USEPA, 1996a) [32] with some modifications. Pretreatment of the soil sample based on “Pretreatment of real samples” and the process was continued as described in “MSPE–DLLME procedure”. The soil samples were spiked with the PAHs standard solutions at two concentration levels (80 and 400 ng kg−1) to assess matrix effects. The RR% and RSD% of analytes were in the range of 62.3–104.3 and 1.3–16.9%, respectively; Fig. 5b shows the chromatograms obtained for soil blank and spiked of 400 ng kg−1 for each of PAHs and as shown in Table 3. It is obvious that this method is applicable to complex environmental samples.
Conclusions
In the present method, for the first time, a highly sensitive solid-phase-dispersive microextraction method based on magnetic carbon nanocomposites coupled with dispersive liquid–liquid microextraction and two miscible stripping solvents (MSPE–DLLME) followed by gas chromatography–mass spectrometry (GC–MS) was developed to the extraction and determination of ultra-trace amounts of 16 PAHs in real environmental samples including tap water, sea water, waste water, sewage, and soil samples. The method provided wide linear ranges, low limits of detection, good precisions, and very high pre-concentration factors for the analysis of environmental samples. Altogether, this combined MSPE–DLLME method is an excellent extraction and determination method for the ultra-trace amounts of PAHs in complex environmental samples, as it is very sensitive, low expensive, effective, and eco-friendly analytical method.
References
Sun F, Littlejohn D, Mark DG (1998) Ultrasonication extraction and solid phase extraction clean-up for determination of US EPA 16 priority pollutant polycyclic aromatic hydrocarbons in soils by reversed-phase liquid chromatography with ultraviolet absorption detection. Anal Chim Acta 364:1–11
Ma J, Xiao R, Li J, Yu J, Zhang Y, Chenb L (2010) Determination of 16 polycyclic aromatic hydrocarbons in environmental water samples by solid-phase extraction using multi-walled carbon nanotubes as adsorbent coupled with gas chromatography–mass spectrometry. J Chromatogr A 1217:5462–5469
Bernal J, Nozal M, Toribio L, Serna M, Borrull F, Marc R (1997) Determination of polycyclic aromatic hydrocarbons in waters by use of supercritical fluid chromatography coupled on-line to solid-phase extraction with disks. Jf Chromatogr A 778:321–328
Filipkowska A, Ludwik L, Grazyna K (2005) Polycyclic aromatic hydrocarbon analysis in different matrices of the marine environmen. Anal Chim Acta 547:243–254
Fladung NC (1995) Optimization of automated solid-phase extraction for quantitation of polycyclic aromatic hydrocarbons in aqueous media by high-performance liquid chromatography–UV detection. J Chromatogr A 692:21–26
Rahimi M, Noroozian E (2014) Frits coated with nano-structured conducting copolymer for solid-phase extraction of polycyclic aromatic hydrocarbons in water samples and liquid chromatographic analysis. Talanta 123:224–232
Shariati-Feizabadi S, Yamini Y, Bahramifar N (2003) Headspace solvent microextraction and gas chromatographic determination of some polycyclic aromatic hydrocarbons in water samples. Anal Chim Acta 489:21–31
Han Q, Wang Z, Xia J, Chen S, Zhang X, Ding M (2012) Facile and tunable fabrication of Fe3O4/graphene oxide nanocomposites and their application in the magnetic solid-phase extraction of polycyclic aromatic hydrocarbons from environmental water samples. Talanta 101:388–395
Liu X, Lu X, Huang Y, Liu C, Zhao S (2014) Fe3O4@ionic liquid@methyl orange nanoparticles as a novel nano-adsorbent for magnetic solid-phase extraction of polycyclic aromatic hydrocarbons in environmental water samples. Talanta 119:341–347
Reyes-Gallardo EM, Lucena R, Cárdenas S, Valcárcel M (2014) Magnetic nanoparticles-nylon 6 composite for the dispersive micro solid phase extraction of selected polycyclic aromatic hydrocarbons from water samples. J Chromatogr A 1345:43–49
Yang N, Zhu S, Zhang D, Xu S (2008) Synthesis and properties of magnetic Fe3O4-activated carbon nanocomposite particles for dye removal. Mater Lett 62:645–657
Yazdanfar N, Yamini Y, Ghambarian M (2014) Homogeneous liquid–liquid microextraction for determination of organochlorine pesticides in water and fruit samples. Chromatographia 77:329–336
Kamankesh M, Mohammadi A, Hosseini H, Modarres Tehrani Z (2015) Rapid determination of polycyclic aromatic hydrocarbons in grilled meat using microwave-assisted extraction and dispersive liquid–liquid microextraction coupled to gas chromatography–mass spectrometry. Meat Sci 103:61–67
Tseng W, Chen P, Huang S (2014) Optimization of two different dispersive liquid–liquid microextraction methods followed by gas chromatography–mass spectrometry determination for polycyclic aromatic hydrocarbons (PAHs) analysis in water. Talanta 120:425–432
Fernández M, Clavijo S, Forteza R, Cerdà V (2015) Determination of polycyclic aromatic hydrocarbons using lab on valve dispersive liquid–liquid microextraction coupled to high performance chromatography. Talanta 138:190–195
Khalili Zanjani MR, Yamini Y, Shariati S, Jonsson JA (2007) A new liquid-phase microextraction method based on solidification of floating organic drop. Anal Chim Acta 585:286–293
Saleh A, Yamini Y, Faraji M, Rezaee M, Ghambarian M (2009) Ultrasound-assisted emulsification microextraction method based on applying low density organic solvents followed by gas chromatography analysis for the determination of polycyclic aromatic hydrocarbons in water samples. J Chromatogr A 1216:6673–6679
Kolahgar B, Hoffmann A, Heiden AC (2002) Application of stir bar sorptive extraction to the determination of polycyclic aromatic hydrocarbons in aqueous samples. J Chromatogr A 963:225–230
Peter P, Bauer C, Wennrich L (2001) Application of stir bar sorptive extraction in combination with column liquid chromatography for the determination of polycyclic aromatic hydrocarbons in water samples. Anal Chim Acta 436:1–9
Doong R, Chang S, Sun Y (2000) Solid-phase microextraction for determining the distribution of sixteen US Environmental Protection Agency polycyclic aromatic hydrocarbons in water samples. J Chromatogr A 879:177–188
Zuazagoitia D, Millán E, Garcia R (2007) A screening method for polycyclic aromatic hydrocarbons determination in water by headspace SPME with GC–FID. Chromatographia 66:773–777
Charalabaki M, Psillakis E, Kalogerakis N, Mantzavinos D (2005) Analysis of polycyclic aromatic hydrocarbons in wastewater treatment plant effluents using hollow fibre liquid-phase microextraction. Chemosphere 60:690–698
Cacho JI, Campillo N, Viñas P, Hernández-Córdoba M (2014) Use of headspace sorptive extraction coupled to gas chromatography–mass spectrometry for the analysis of volatile polycyclic aromatic hydrocarbons in herbal infusions. J Chromatogr A 1356:38–44
Shamsipur M, Yazdanfar N, Ghambarian M (2016) Combination of solid-phase extraction with dispersive liquid–liquid microextraction followed by GC–MS for determination of pesticide residues from water, milk, honey and fruit juice. Food Chem 204:289–297
Mehdinia A, Khojasteh E, Baradaran Kayyal T, Jabbari A (2014) Magnetic solid phase extraction using gold immobilized magnetic mesoporous silica nanoparticles coupled with dispersive liquid–liquid microextraction for determination of polycyclic aromatic hydrocarbons. J Chromatogr A 1364:20–27
López-Jiménez F, Ballesteros-Gómez A, Rubio S (2014) Determination of polycyclic aromatic hydrocarbons (PAH4) in food by vesicular supramolecular solvent-based microextraction and LC–fluorescence detection. Food Chem 143:341–347
Aragón Á, Toledano R, Vázquez A, Villén J, Cortés J (2015) Analysis of polycyclic aromatic hydrocarbons in aqueous samples by large volume injection gas chromatography–mass spectrometry using the through oven transfer adsorption desorption interface. Talanta 139:1–5
Pincemaille J, Schummer C, Heinen E, Moris G (2014) Determination of polycyclic aromatic hydrocarbons in smoked and non-smoked black teas and tea infusions. Food Chem 145:807–813
Tajabadi F, Yamini Y, Sovizi MR (2013) Carbon-based magnetic nanocomposites in solid phase dispersion for the preconcentration some of lanthanides, followed by their quantitation via ICP-OES. Microchim Acta 180:65–73
Guo L, Kee Lee H (2011) Low-density solvent-based solvent demulsification dispersive liquid–liquid microextraction for the fast determination of trace levels of sixteen priority polycyclic aromatic hydrocarbons in environmental water samples. J Chromatogr A 1218:5040–5046
Rezaee M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadia F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid–liquid microextraction. J Chromatogr A 1116:1–9
Simpson SA, Burston LB, Jolley DF, Chau K (2006) Application of surrogate methods for assessing the bioavailability of PAHs in sediments to a sediment ingesting bivalve. Chemosphere 65:2401–2410
Acknowledgements
The authors greatly appreciate the support of this work by the Research Councils of Razi University and Iranian Research and Development Center for Chemical Industries and Iran National Elite Foundation (INEF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human or animal subjects.
Informed consent
Informed consent was not applicable.
Rights and permissions
About this article

Cite this article
Yazdanfar, N., Shamsipur, M., Ghambarian, M. et al. A Highly Sensitive Dispersive Microextraction Method with Magnetic Carbon Nanocomposites Coupled with Dispersive Liquid–Liquid Microextraction and Two Miscible Stripping Solvents Followed by GC–MS for Quantification of 16 PAHs in Environmental Samples. Chromatographia 81, 487–499 (2018). https://doi.org/10.1007/s10337-018-3469-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-018-3469-5