
Abstract
A new, sensitive and fast high-performance liquid chromatography–diode-array detection assay is herein reported, for the first time, to simultaneously quantify carbamazepine (CBZ), oxcarbazepine (OXC), and the active metabolites carbamazepine-10,11-epoxide (CBZ-E) and licarbazepine (LIC) in HepaRG cell culture medium samples. Chromatographic separation of analytes (CBZ, CBZ-E, OXC, LIC) and internal standard (IS) was achieved in less than 15 min on a C18-column, at 35 °C, using a mobile phase composed of water/methanol/acetonitrile (69:25:6 v/v/v) pumped at 1 mL min−1. The analytes and IS were detected at 215 nm. The method proved to be selective, accurate (bias ± 14.6 %), precise (coefficient of variation ≤13.1 %) and linear (r 2 ≥ 0.9901) over the concentration ranges of 0.1–15 μg mL−1 for CBZ; 0.1–5 μg mL−1 for CBZ-E and OXC; and 0.1–40 μg mL−1 for LIC. Furthermore, the absolute recovery of the analytes ranged from 64.5 to 96.9 % and their stability was demonstrated in the studied conditions. This validated HPLC assay will be a suitable tool to support future in vitro metabolism profiling, drug interaction and other pharmacokinetic-based studies in HepaRG cells involving these antiepileptic drugs (CBZ and OXC) and their main metabolites.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epilepsy is one of the most common serious chronic neurological disorders, affecting 50 million people worldwide [1–3]. It is characterized by recurrent unprovoked seizures and the main goal of epilepsy treatment is the achievement of a complete seizure-freedom without deleterious effects. For that, a continuous antiepileptic activity is required, which will be achieved only if plasma and brain drug concentrations remain continuously at therapeutic levels [4].
Since the beginning of the 1990s several new antiepileptic drugs (AEDs) were approved by the Food and Drug Administration and/or by the European Medicines Agency [e.g., eslicarbazepine acetate, felbamate, gabapentin, lamotrigine, levetiracetam, oxcarbazepine (OXC), pregabalin, rufinamide, stiripentol, tiagabine, topiramate, vigabatrin and zonisamide]. The majority of these AEDs offers appreciable advantages in terms of their favourable pharmacokinetics, improved tolerability and lower potential for drug interactions comparatively to the older AEDs [e.g., phenobarbital, phenytoin, ethosuximide, carbamazepine (CBZ), and valproic acid] [5, 6]. However, despite the large therapeutic arsenal of old and new AEDs, approximately 55–60 % of people with focal epilepsy and up to 20 % of patients with primary generalized epilepsy develops drug resistance [7]. Hence, many patients are not seizure-free even under appropriate pharmacotherapy with the currently available AEDs, including under combination therapy with rational polytherapy regimens [5]. Thus, the search for novel AEDs with better efficacy and a safer therapeutic index is clearly warranted [5–8].
CBZ and OXC are clinically well-established AEDs, presenting a substantial overlap in terms of pharmacological and therapeutic properties [9]. CBZ and OXC are structurally related drugs that share the dibenzazepine nucleus but they differ at the 10-,11-position (Fig. 1). These molecular differences are possibly on the basis of their different metabolic profiles. In humans, whereas CBZ undergoes oxidative metabolism to carbamazepine-10,11-epoxide (CBZ-E), OXC is rapidly reduced to its pharmacologically active licarbazepine (LIC) metabolite (Fig. 1) [10–12].

Metabolic pathways of carbamazepine (CBZ) and oxcarbazepine (OXC) to their active metabolites carbamazepine-10,11-epoxide (CBZ-E) and licarbazepine (LIC), respectively
Although clinical benefits are not expected with long-term CBZ and OXC cotherapy, switching from CBZ to OXC is a common practice in patients unsatisfactorily treated with CBZ [13–15]. Nevertheless, these drugs are commonly used in combination therapy regimens with other AEDs in refractory patients [16–18]; actually, when monotherapy fails, add-on therapy is an alternative option and there are many possible AED combinations based on their mechanisms of action and pharmacokinetic interactions. Nowadays, polytherapy regimens with AEDs are prevalent and the potential for AEDs to interact results in many challenges [18, 19]. These concerns were recently reviewed by Landmark and Patsalos [19] and the importance of the availability of appropriate in vitro models to characterize potential interactions involving AEDs was also highlighted.
Accordingly, as CBZ and OXC are widely used in the clinical practice and taking into account the valuable characteristics of the HepaRG cell line, which constitutes a new and promising alternative model to the primary human hepatocytes (“gold standard” model) for the evaluation of the hepatic drug metabolism in in vitro conditions [20], this work was planned to make available a useful bioanalytical method to support future in vitro pharmacokinetic-based studies in cultured HepaRG cells involving CBZ and/or OXC. Among other advantages, HepaRG cells possess the metabolic capacity characteristic of primary human hepatocytes and the indefinite proliferation property of hepatoma cell lines, constituting a promising in vitro tool to assess the drug metabolite profiling, the hepatic kinetics of drugs and to foresee drug–drug interactions [21–23]. In fact, the use of in vitro data for the assessment of potential drug–drug interactions is also a practice widely accepted by the regulatory authorities in the development of new drugs [24].
Hence, to the best of our knowledge, it is herein reported for the first time a high-performance liquid chromatography method with diode-array detection (HPLC–DAD) to quantify CBZ, OXC, and their pharmacologically active metabolites (CBZ-E and LIC, respectively) in HepaRG cell culture samples.
Materials and Methods
Reagents and Cells
CBZ, CBZ-E, OXC and primidone (PRM), used as internal standard (IS), were purchased from Sigma–Aldrich (St Louis, MO, USA). LIC was supplied by Tocris Bioscience (Bristol, UK). Methanol and acetonitrile, both of HPLC gradient grade, were purchased from Fisher Scientific (Leicestershire, UK) and the ultra-pure water (HPLC grade, >18 MΩ cm) was prepared by means of a Milli-Q water apparatus from Millipore (Milford, MA, USA). Ethyl acetate were purchased from Fisher Scientific (Leicestershire, UK). HepaRG cells (lot no. #48588) were obtained from Life Technologies—Invitrogen™ (through Alfagene, Portugal). All cell culture reagents including Williams’ E medium, foetal bovine serum, hydrocortisone hemisuccinate, dimethyl sulfoxide and trypsin–EDTA were purchased from Sigma–Aldrich (St Louis, MO, USA).
Stock Solutions, Calibration Standards and Quality Control Samples
Stock solutions of CBZ (15 mg mL−1), CBZ-E (30 mg mL−1), OXC (10 mg mL−1) and LIC (10 mg mL−1) were individually prepared by dissolving the appropriate amount of each compound in methanol. These solutions were then adequately diluted in water–methanol (50:50, v/v) to obtain the corresponding working solutions. Afterwards, stock and working solutions of drugs and metabolites were properly mixed to afford six combined spiking solutions with final concentrations 1, 2, 6, 20, 80 and 150 μg mL−1 for CBZ; 1, 2, 5, 10, 30 and 50 μg mL−1 for CBZ-E and OXC; and 1, 2, 10, 50, 250 and 400 μg mL−1 for LIC. Each one of these combined solutions was daily used for spiking aliquots of blank supplemented Williams’ E medium to prepare six calibration standards in the concentration ranges of 0.1–15 μg mL−1 for CBZ; 0.1–5 μg mL−1 for CBZ-E and OXC; and 0.1–40 μg mL−1 for LIC. Regarding the IS, the stock solution was also prepared in methanol (2 mg mL−1) and the working solution (500 μg mL−1) was obtained through the dilution of an appropriate volume of the stock solution with water–methanol (50:50, v/v). With exception of the IS working solution which was daily prepared, all the stock, working and combining solutions were stored at 4 °C and protected from light.
Quality control (QC) samples at three representative concentration levels, representing the low (QC1), medium (QC2) and high (QC3) ranges of the calibration curves, were also independently prepared in supplemented Williams’ E medium. With that purpose, aliquots of blank supplemented Williams’ E medium were spiked to attain final concentrations of 0.3, 7.5 and 13.5 μg mL−1 for CBZ; 0.3, 2.5 and 4.5 μg mL−1 for CBZ-E and OXC; and 0.3, 20 and 36 μg mL−1 for LIC. One additional QC sample was also prepared at the concentration of the lower limit of quantification (QCLLOQ).
Apparatus and Chromatographic Conditions
Chromatographic analysis was carried out using an HPLC system (Shimadzu LC-2010A HT Liquid Chromatography) coupled with DAD (Shimadzu SPD-M20A). All instrumental parts were automatically controlled by LabSolutions software (Shimadzu, Kyoto, Japan). The chromatographic separation of CBZ, CBZ-E, OXC, LIC and IS was carried out at 35 °C on a reversed-phase LiChroCART® Purospher Star column (C18, 55 × 4 mm; 3 µm particle size) purchased from Merck KGaA (Darmstadt, Germany). An isocratic elution was applied at a flow rate of 1.0 mL min−1 with a mobile phase composed of water/methanol/acetonitrile (69:25:6 v/v/v). The mobile phase was filtered through a 0.2 µm filter and degassed ultrasonically for 15 min before use. The injection volume was 20 µL and the wavelength of 215 nm was selected for the detection of all compounds.
Sample Preparation and Extraction
Each aliquot (200 µL) of supplemented Williams’ E culture medium was added with 20 µL of IS working solution, 300 µL of acetonitrile and then with 1 mL of ethyl acetate. Afterwards, the mixture was vortex-mixed for 30 s and centrifuged at 13,500 rpm (3 min). The organic layer was transferred to a glass tube and the aqueous layer was re-extracted twice more with ethyl acetate (1 mL each time) using the conditions previously described. The combined organic phases were evaporated to dryness under a gentle nitrogen stream at 45 °C and then reconstituted with 100 µL of mobile phase. An aliquot (20 µL) of this final sample was injected into the chromatographic system.
Method Validation
The developed method was appropriately validated taking into account the international accepted recommendations for bioanalytical method validation [25–27]. The validation was carried out considering the acceptance criteria proposed for specific validation parameters including selectivity, linearity, precision, accuracy, lower limit of quantification (LLOQ), recovery and stability.
Selectivity
Aiming at testing the chromatographic similarity between the supplemented Williams’ E medium collected after the culture of HepaRG cells, to reproduce in the best way what happens in real metabolic studies, and the simple supplemented Williams’ E medium, a set of samples from these two related matrices was analysed and compared. To obtain aliquots of medium from cultured HepaRG cells the following procedures were conducted. HepaRG cells were maintained in the Williams’ E medium supplemented with 10 % foetal bovine serum, 100 IU mL−1 penicillin, 100 µg mL−1 streptomycin, 5 µg mL−1 insulin, 2 mM glutamine and 5 × 10−5 M hydrocortisone hemisuccinate during 15 days. After this period of time the confluence was reached and cells were then maintained in the same culture medium but supplemented with 2 % dimethyl sulfoxide (differentiation medium) for more 15 days to obtain differentiated HepaRG cell cultures. It is worthy to mention that HepaRG cell cultures were maintained at 37 °C in humidified air incubator with 5 % CO2 and the medium was renewed each 3 days. Then, the differentiated HepaRG cells were seeded at a high density (4.5 × 105 cells cm−2) in 96-well plates and maintained during 48 h. After that, the cells were exposed to 200 µL of supplemented Williams’ E medium for 12 h and then the total volume of each well was collected and 300 µL of acetonitrile was immediately added.
Calibration Curve
The linearity of this chromatographic assay was evaluated for each analyte within the concentration ranges defined in “Stock solutions, calibration standards and quality control samples”. For this purpose, calibration curves were prepared using six spiked supplemented Williams’ E medium calibration standards and assayed on three different days (n = 3). Calibration curves were constructed by plotting the analyte–IS peak area ratio as function of the corresponding nominal concentrations. The data were fitted to a weighted linear regression analysis and the weighting factor that yields the best fit of peak–area ratios versus concentration is selected [28].
Lower Limit of Quantification
The LLOQ, defined as the lowest concentration of the calibration curve which can be measured with adequate inter- and intraday precision, and accuracy [26, 27] was evaluated by analysing supplemented Williams’ E medium samples prepared in replicates. The precision, expressed as percentage of the coefficient of variation (CV), cannot exceed 20 %; whereas accuracy, expressed by the deviation from nominal concentration value (bias), should be within ±20 %.
Precision and accuracy
QC samples analysed on three consecutive days (n = 3) at the four concentration levels (QCLLOQ, QC1, QC2 and QC3) representative of the calibration range were used to investigate the interday precision and accuracy of the assay. Similarly, the intraday precision and accuracy were also assessed by analysing five sets of QC samples in a single day (n = 5). According to the acceptance criteria defined by the bioanalytical method validation guidelines the intra- and interday precision (expressed as percentage of CV) should be lower than or equal to 15 % (or 20 % in the LLOQ), and the intra- and interday accuracy (expressed as percentage of bias) should be within ±15 % (or ±20 % in the LLOQ) [26, 27].
Recovery
The absolute recovery of the analytes from the samples submitted to the treatment previously described in the “Sample Preparation and Extraction” was determined using three QC samples (QC1, QC2 and QC3). The recovery was calculated comparing the analytes peak areas from extracted QC supplemented Williams’ E medium samples with those obtained after direct injection of non-extracted solutions at the same nominal concentrations (n = 5). Similarly, the recovery of IS was also evaluated at the concentration used in sample analysis, calculating the ratio of its peak areas in extracted samples and non-extracted solutions.
Stability
The stability of the analytes in supplemented Williams’ E medium was investigated for QC1 and QC3 (n = 5). The data of the QC samples analysed before (reference samples) were compared with those obtained after being exposed to the experimental conditions for stability assessment (stability samples). As stability criterion (n = 5), a stability/reference samples ratio of 85–115 % was accepted [27, 31]. The short- and long-term stability were evaluated, respectively, at room temperature for 4 h and −20 °C for 8 days (n = 5), aiming at simulating sample handling and storage time in the freezer before analysis. Moreover, in an attempt to simulate the time that samples can remain in the auto-sampler before analysis, the post-preparative stability of the analytes in processed samples was also studied at room temperature during 12 h.
Results
Method Validation
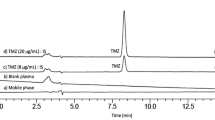
The chromatographic separation of CBZ, CBZ-E, OXC and LIC in spiked supplemented Williams’ E medium culture samples was successfully achieved using the chromatographic conditions previously described. Under these analytical conditions the last-eluting analyte was CBZ, being the run time of approximately 15 min. The order of elution of the compounds was the following: PRM (IS), LIC, CBZ-E, OXC and CBZ. Representative chromatograms of blank and spiked supplemented Williams’ E culture medium samples are shown in Fig. 2.

Typical chromatograms of extracted supplemented Williams’ E medium samples generated by the HPLC–DAD assay developed: blank supplemented Williams’ E medium (a); supplemented Williams’ E medium spiked with internal standard (IS) and the analytes at concentrations of the lower limit of quantification (b); and supplemented Williams’ E medium spiked with IS and the analytes at intermediate concentrations of the calibration ranges (c). CBZ carbamazepine; CBZ-E carbamazepine-10,11-epoxide; PRM primidone; LIC licarbazepine; OXC oxcarbazepine
Selectivity
The chromatograms obtained by the analysis of blank samples of supplemented Williams’ E medium collected after the culture of HepaRG cells (to reproduce the metabolic studies) and those generated by the analysis of blank samples of the simple supplemented Williams’ E medium were broadly comparable. As result, the most convenient blank matrix (the simple supplemented Williams’ E medium) was chosen for the development and validation of this HPLC–DAD assay.
Calibration Curves and LLOQs
The calibration curves obtained in supplemented Williams’ E culture medium were linear (r 2 ≥ 0.9901; Table 1) for all the analytes over the concentration range defined in “Stock Solutions, Calibration Standards and Quality Control Samples” and showed a consistent correlation between analyte–IS peak area ratios and the corresponding nominal concentrations. The calibration curves for all the analytes were subjected to weighted linear regression analysis using 1/x 2 as the weighting factor, due to the wide calibration range and to compensate for heteroscedasticity detected. This factor was chosen taking into account the plots and the sums of absolute percentage of relative error as statistical criteria. The regression equations of the calibration curves and the corresponding determination coefficients (r 2) achieved for each analyte are summarized in Table 1.
The LLOQs were experimentally defined as 0.1 μg mL−1 for all the analytes (CBZ, CBZ-E, OXC and LIC) with acceptable precision (CV ≤17.3 %) and accuracy (bias varied from −12.3 to 10.1 %).
Precision and Accuracy
The data for intra- and interday precision and accuracy obtained from QC supplemented Williams’ E medium samples at the four different concentration levels (QCLLOQ, QC1, QC2 and QC3) are shown in Table 2. All the data fulfilled the acceptance criteria established. Accordingly, the overall inter- and intraday CV values did not exceed 13.1 % (or 17.7 % in the QCLLOQ), and the overall inter- and intraday bias values varied between −12.8 and 14.6 % (or −12.3–10.1 % in the QCLLOQ), which supports that the HPLC–DAD method herein described is precise and accurate.
Recovery
The overall absolute recovery of CBZ, CBZ-E, OXC and LIC from supplemented Williams’ E culture medium samples was determined at three concentration levels (QC1, QC2 and QC3) by repeated analysis (n = 5). These data are shown in Table 3. The mean absolute recoveries ranged from 64.5 to 96.9 % and showed CV values lower than 14.8 % for all analytes (AEDs and metabolites). The recovery of the IS (PRM) was also evaluated, being its absolute recovery of 70.2 ± 8.6 % with a CV value of 12.2 %. These data undoubtedly support a consistent recovery over the evaluated concentration ranges by the sample preparation procedure implemented.
Stability
The stability of the CBZ, CBZ-E, OXC and LIC in supplemented Williams’ E culture medium was evaluated under the different conditions previously described, which simulate the handling and sample storage conditions to be likely required during the analytical process. The analytes demonstrated to be stable in unprocessed samples at room temperature for 4 h and at −20 °C for 8 days and also in processed samples at room temperature during 12 h. The stability data are shown in Table 4.
Method Application
HepaRG cells were seeded in Williams’ E medium supplemented with 10 % foetal bovine serum, 100 IU mL−1 penicillin, 100 μg mL−1 streptomycin, 5 μg mL−1 insulin, 2 mM glutamine and 5 × 10−5 M hydrocortisone hemisuccinate (basal growth medium) and maintained at 37 °C in a humidified air incubator with 5 % CO2. The medium was renewed every 2–3 days until cells reach confluence, which were expanded by gentle trypsinization and maintained in culture by serial passages and then used when appropriate for the specific studies. For the metabolic studies, HepaRG cells were maintained in the basal growth medium during 15 days. After this period of time the confluence was reached and the cells were maintained in the same culture medium supplemented with 2 % DMSO (differentiation medium) for more 15 days to obtain differentiated HepaRG cell cultures. In both cases, cell cultures were maintained at 37 °C in humidified air incubator with 5 % CO2 and the medium was renewed each 3 days. The differentiated HepaRG cells were then seeded at a high density (4.5 × 105 cells cm−2) on 96-well plates. After a period of cell adhesion of 24 h, the cells were incubated with 200 µL OXC 2.5 μg mL−1 prepared in foetal bovine serum-free basal growth medium (0.25 % DMSO). After 2, 4, 6, 12 and 24 h, the culture medium (200 µL) was collected and 300 µL of ice-cold acetonitrile was added to each sample to stop the metabolic reactions. During the incubation times, the cell cultures were maintained at 37 °C in humidified air incubator with 5 % CO2. Thereafter, the samples were frozen at −20 °C until analysis of OXC and its metabolite (LIC). The obtained results are presented in Fig. 3a, showing an increasing formation of LIC over the time while the concentration of OXC (parent drug) is gradually reduced. In Fig. 3b is shown a chromatogram resulting of the analysis of a real sample obtained at 6 h after incubation of OXC (2.5 μg mL−1) with the HepaRG cells.

Concentration-time profiles for the metabolism of OXC in its active metabolite LIC after 2, 4, 6, 12 and 24 h of incubation of the parent drug (OXC) at 2.5 μg mL−1 in differentiated HepaRG cells (a); and a representative chromatogram of the analysis of a real sample obtained after 6 h of incubation of OXC (2.5 μg mL−1) in the differentiated HepaRG cells (b). OXC oxcarbazepine, LIC licarbazepine, PRM primidone
Discussion
As previously mentioned, besides the availability of more than 20 AEDs [1], the truth is that the monotherapy with AEDs often fails, requiring polytherapy regimens in an attempt to achieve better seizure control and fewer side effects [29–32]. Unfortunately, the implementation of polytherapy regimens in the clinical practice frequently originates complex and unpredictable pharmacokinetic and pharmacodynamic interactions, leading to possible clinical consequences in terms of toxicity or even therapeutic inefficacy [30, 33]. In fact, patients with epilepsy are particularly affected by clinically relevant drug interactions [33–35]. This can be explained not only by the long administration periods of AEDs, often required for a lifetime, increasing the probability of coprescription, but also due to the narrow therapeutic index of some AEDs (e.g., phenobarbital, phenytoin, CBZ, valproic acid); in fact, some of these older AEDs (e.g., CBZ, valproic acid) are still widely prescribed today and have marked effects on the activity of cytochrome P450 (CYP) isoenzymes, which also metabolize the majority of existing medication, including the new generation AEDs [33]. Although other drug interaction mechanisms can occur, such as pharmacodynamic-based interactions, the majority of clinically significant AED interactions are result of induction or inhibition of drug-metabolizing CYP isoenzymes [33]. The enzyme inducers as CBZ, phenytoin and phenobarbital increase the activity of a variety of CYP isoenzymes (CYP1A2, CYP2C9, CYP2C19 and CYP3A4), as well as glucuronyl transferases and epoxide hydrolase, which may enhance the drug metabolism and reduce the serum concentrations of other concurrently administered drugs (including AEDs) [33, 34, 36]. Actually, although none of the newer AEDs shares the broad spectrum enzyme-inducing activity of those older generation agents, they are also cleared fully or partly by inducible CYP isoenzymes, making them a target for drug interactions mediated by enzyme induction [33]. On the other hand, several drugs from other therapeutic classes have also been found to increase the serum concentration of AEDs, presumably by inhibiting their metabolism. For example, several antidepressants like fluoxetine, fluvoxamine or trazodone and antimicrobials like clarithromycin, erythromycin or fluconazole have been reported to interfere in the serum concentrations of CBZ [33]. Consequently, whenever possible, these drug interactions should be prevented and early identified [35]. Indeed, drug–drug interactions are an essential aspect to be considered in the process of new drug development, representing a major concern for pharmaceutical industry and regulatory agencies and at clinical level for healthcare professionals and their patients. Therefore, in vitro models are being increasingly used during preclinical drug development, arising as screening tools to predict drug–drug interactions [37–40]. In fact, there is increasing evidence that appropriate in vitro drug interaction studies can accurately reflect what happens in in vivo conditions [37, 39]. Indeed, in the decision making process during the development of new drug candidates, the evaluation of induction and inhibition of CYP isoenzymes is one of the major points of concern [41, 42]. Although in the past years numerous in vitro and/or ex vivo models have been developed to investigate drug metabolism, the HepaRG cells represent perhaps nowadays the most relevant in vitro model as surrogate to the primary human hepatocytes, evidencing a great value to foresee drug–drug interactions [22, 23].
Several metabolic interactions involving AEDs have been identified by means of in vitro models. For instance, it is the case of the metabolic interaction between imipramine [43] or felbamate [44] with CBZ. Taking into account the experimental advantages of HepaRG cells, the availability of a rapid, sensitive and reliable analytical method is critical to support subsequent metabolic drug interaction studies in this kind of biological samples. Up to date, several HPLC methods have been developed and validated for the determination of these AEDs, and some of their main metabolites in several matrices. Most of the methodologies described in the literature for the simultaneous determination of CBZ and OXC are HPLC assays using DAD or ultraviolet detection [30, 45–50]. Nevertheless, the extraction procedures that were applied in the preparation of the samples were diversified and included not only the liquid–liquid extraction [51], that was also applied in this assay, but also solid-phase extraction [30, 45, 47, 49], microextraction by packed sorbent [46, 50] and protein precipitation [48]. The matrices used in these methodologies included human plasma [30, 45–48, 50] or urine [50], for example. However, none of the HPLC assays described permitted the quantification of CBZ or/and OXC, and their main metabolites, in culture mediums or even in supernatants of cell cultures. Actually, the HPLC–DAD assay herein described is the first one to quantify CBZ, OXC, and their relevant pharmacologically active metabolites (CBZ-E and LIC, respectively) in HepaRG cell culture medium.
During the process of drug discovery and development the bioanalysis was recognised to be a critical tool, essential for the pharmacokinetic/pharmacodynamics characterization of a drug. Indeed, there is a continuous need of development of analytical methodologies, which permit the quantification of not only the drug but also its metabolites in several biological samples, supporting various stages of the drug discovery and development [52, 53]. However, it is accepted that in the latter stages of the process the levels of analytical acceptance criteria become stricter, and it is supposed that the results were confirmed by appropriate validation assays, which permit to attest the reliability, robustness and accuracy of the methods [52–54]. Nevertheless, the degree of development and quality of bioanalytical assays used in the discovery and development of novel chemical entities tends to increase as the lead candidates progress to more advanced stages. Actually, the various validation criteria defined by the international guidelines are not usually applied in the early stages of drug discovery and development, being used only minimal standards in the higher throughput assays performed in these steps [52–54]. All these points could explain the lack of HPLC methodologies in the literature describing a full bioanalytical method validation regarding the in vitro studies. Nonetheless, as we intended to develop a robust technique that could provide a high level of confidence in the results obtained, the HPLC–DAD method herein described was extensively validated taking into account the international criteria of the US Food and Drug Administration (FDA) and by the European Medicines Agency (EMA) guidelines.
Conclusion
Until now, to the best of our knowledge, there is no reported methodology in the literature to simultaneously determine the considered analytes CBZ, CBZ-E, OXC and LIC in HepaRG cell culture medium samples. Therefore, the present work describes the first HPLC–DAD assay developed and fully validated according to the international requirements for bioanalytical purposes. Hence, this methodology represents a useful bioanalytical tool to support future in vitro metabolic, drug interaction and other pharmacokinetic-based studies involving these AEDs and metabolites in the new and promising in vitro model HepaRG cell line.
References
Zhang C, Kwan P, Zuo Z, Baum L (2012) The transport of antiepileptic drugs by p-glycoprotein. Adv Drug Deliv Rev 64:930–942
Galanopoulou AS, Buckmaster PS, Staley KJ et al (2012) Identification of new epilepsy treatments: issues in preclinical methodology. Epilepsia 53:571–582
Perucca E (2001) Clinical pharmacology and therapeutic use of the new antiepileptic drugs. Fundam Clin Pharmacol 15:405–417
Schmidt D (2009) Drug treatment of epilepsy: options and limitations. Epilepsy Behav 15:56–65
Brodie MJ (2010) Antiepileptic drug therapy the story so far. Seizure 19:650–655
Bialer M (2012) Chemical properties of antiepileptic drugs (AEDs). Adv Drug Deliv Rev 64:887–895
Weaver DF (2013) Design of innovative therapeutics for pharmacoresistant epilepsy: challenges and needs. Epilepsia 54(Suppl 2):56–59
Weaver DF, Pohlmann-Eden B (2013) Pharmacoresistant epilepsy: unmet needs in solving the puzzle(s). Epilepsia 54(Suppl 2):80–85
French JA, Gazzola DM (2011) New generation antiepileptic drugs: what do they offer in terms of improved tolerability and safety? Ther Adv Drug Saf 2:141–158
Benes J, Parada A, Figueiredo AA et al (1999) Anticonvulsant and sodium channel-blocking properties of novel 10,11-dihydro-5H-dibenz[b, f]azepine-5-carboxamide derivatives. J Med Chem 42:2582–2587
Flesch G (2004) Overview of the clinical pharmacokinetics of oxcarbazepine. Clin Drug Investig 24:185–203
Alves G, Fortuna A, Sousa J et al (2010) Enantioselective assay for therapeutic drug monitoring of eslicarbazepine acetate: no interference with carbamazepine and its metabolites. Ther Drug Monit 32:512–516
Albani F, Baruzzi A (2006) Oxcarbazepine long-term treatment retention in patients switched over from carbamazepine. Neurol Sci 27:173–175
Vaisleib II, Neft RA (2008) Overnight transition from carbamazepine to oxcarbazepine in children and adolescents. Pharmacotherapy 28:1211–1214
Lee SA, Heo K, Kim WJ et al (2010) Clinical feasibility of immediate overnight switching from slow-release carbamazepine to oxcarbazepine in Korean patients with refractory partial epilepsy. Seizure 19:356–358
Guberman A (1998) Monotherapy or polytherapy for epilepsy? Can J Neurol Sci 25:S3–S8
French JA, Faught E (2009) Rational polytherapy. Epilepsia 50(Suppl 8):63–68
Giussani G, Beghi E (2013) Does mechanism of drug action matter to inform rational polytherapy in epilepsy? CNS Neurol Disord: Drug Targets 12:426–435
Johannessen Landmark C, Patsalos PN (2012) Methodologies used to identify and characterize interactions among antiepileptic drugs. Expert Rev Clin Pharmacol 5:281–292
Andersson TB (2010) The application of HepRG cells in evaluation of cytochrome P450 induction properties of drug compounds. Methods Mol Biol 640:375–387
Guillouzo A (2008) New perspectives in the use of human hepatocytes in the preclinical drug development process. Ann Pharm Fr 66:288–295
Anthérieu S, Chesné C, Li R et al (2010) Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab Dispos 38:516–525
Darnell M, Schreiter T, Zeilinger K et al (2011) Cytochrome P450-dependent metabolism in HepaRG cells cultured in a dynamic three-dimensional bioreactor. Drug Metab Dispos 39:1131–1138
Walsky RL, Boldt SE (2008) In vitro cytochrome P450 inhibition and induction. Curr Drug Metab 9:928–939
Shah VP, Midha KK, Findlay JW et al (2000) Bioanalytical method validation–a revisit with a decade of progress. Pharm Res 17:1551–1557
US Food and Drug Administration (2001) Guidance for Industry: bioanalytical method validation. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070107.pdf. Accessed 15 Jun 2015
European Medicines Agency (2011) Guideline on bioanalytical method validation. http://www.ema.europa.eu/docs/enGB/documentlibrary/Scientificguideline/2011/08/WC500109686.pdf. Accessed 15 Jun 2015
Almeida AM, Castel-Branco MM, Falcão AC (2002) Linear regression for calibration lines revisited: weighting schemes for bioanalytical methods. J Chromatogr B Anal Technol Biomed Life Sci 774:215–222
Deckers CL, Czuczwar SJ, Hekster YA et al (2000) Selection of antiepileptic drug polytherapy based on mechanisms of action: the evidence reviewed. Epilepsia 41:1364–1374
Bugamelli F, Sabbioni C, Mandrioli R et al (2002) Simultaneous analysis of six antiepileptic drugs and two selected metabolites in human plasma by liquid chromatography after solid-phase extraction. Anal Chim Acta 472:1–10
Fortuna A, Bicker J, Alves G et al (2011) A chiral HPLC-UV method for the quantification of dibenz[b, f]azepine-5-carboxamide derivatives in mouse plasma and brain tissue: eslicarbazepine acetate, carbamazepine and main metabolites. J Sep Sci 34:1391–1401
Garnett WR, St Louis EK, Henry TR, Bramley T (2009) Transitional polytherapy: tricks of the trade for monotherapy to monotherapy AED conversions. Curr Neuropharmacol 7:83–95
Perucca E (2006) Clinically relevant drug interactions with antiepileptic drugs. Br J Clin Pharmacol 61:246–255
Johannessen SI, Landmark CJ (2010) Antiepileptic drug interactions-principles and clinical implications. Curr Neuropharmacol 8:254–267
Riva R, Albani F, Contin M, Baruzzi A (1996) Pharmacokinetic interactions between antiepileptic drugs. Clinical considerations. Clin Pharmacokinet 31:470–493
Brodie MJ, Mintzer S, Pack AM et al (2013) Enzyme induction with antiepileptic drugs: cause for concern? Epilepsia 54:11–27
Lin JH, Lu AY (1997) Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol Rev 49:403–449
Fabre G, Combalbert J, Berger Y, Cano JP (1990) Human hepatocytes as a key in vitro model to improve preclinical drug development. Eur J Drug Metab Pharmacokinet 15:165–171
Costa A, Sarmento B, Seabra V (2014) An evaluation of the latest in vitro tools for drug metabolism studies. Expert Opin Drug Metab Toxicol 10:103–119
Wienkers LC, Heath TG (2005) Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov 4:825–833
McGinnity DF, Riley RJ (2001) Predicting drug pharmacokinetics in humans from in vitro metabolism studies. Biochem Soc Trans 29:135–139
Anthérieu S, Chesné C, Li R et al (2012) Optimization of the HepaRG cell model for drug metabolism and toxicity studies. Toxicol Vitr 26:1278–1285
Daniel W, Netter KJ (1988) Metabolic interaction between imipramine and carbamazepine in vivo and in vitro in rats. Naunyn Schmiedebergs Arch Pharmacol 337:105–110
Egnell AC, Houston B, Boyer S (2003) In vivo CYP3A4 heteroactivation is a possible mechanism for the drug interaction between felbamate and carbamazepine. J Pharmacol Exp Ther 305:1251–1262
Fortuna A, Sousa J, Alves G et al (2010) Development and validation of an HPLC-UV method for the simultaneous quantification of carbamazepine, oxcarbazepine, eslicarbazepine acetate and their main metabolites in human plasma. Anal Bioanal Chem 397:1605–1615
Ferreira A, Rodrigues M, Oliveira P et al (2014) Liquid chromatographic assay based on microextraction by packed sorbent for therapeutic drug monitoring of carbamazepine, lamotrigine, oxcarbazepine, phenobarbital, phenytoin and the active metabolites carbamazepine-10,11-epoxide and licarbazepine. J Chromatogr B Analyt Technol Biomed Life Sci 971:20–29
Franceschi L, Furlanut M (2005) A simple method to monitor plasma concentrations of oxcarbazepine, carbamazepine, their main metabolites and lamotrigine in epileptic patients. Pharmacol Res 51:297–302
Khoschsorur GA, Frühwirth F, Halwachs-Baumann G (2001) Simple and rapid HPLC method for simultaneous determination of multiple antiepileptic drugs in human serum. Chromatographia 54:345–349
Serralheiro A, Alves G, Fortuna A et al (2013) First HPLC-UV method for rapid and simultaneous quantification of phenobarbital, primidone, phenytoin, carbamazepine, carbamazepine-10,11-epoxide, 10,11-trans-dihydroxy-10,11-dihydrocarbamazepine, lamotrigine, oxcarbazepine and licarbazepine in human plas. J Chromatogr B Anal Technol Biomed Life Sci 925:1–9
Rani S, Malik AK, Singh B (2012) Novel micro-extraction by packed sorbent procedure for the liquid chromatographic analysis of antiepileptic drugs in human plasma and urine. J Sep Sci 35:359–366
Meyler M, Kelly MT, Smyth MR (1993) New method for the determination of four antiepileptic drugs in human plasma by high performance liquid chromatography. Chromatographia 36:27–32
Pandey S, Pandey P, Tiwari G, Tiwari R (2010) Bioanalysis in drug discovery and development. Pharm Methods 1:14–24
Korfmacher WA (2011) Bioanalytical support for both in vitro and in vivo assays across drug discovery and drug development. Encyclopedia of Drug Metabolism and Interactions, vol 6, 1st edn. Wiley, pp 1–18
Srinivas NR (2006) Applicability of bioanalysis of multiple analytes in drug discovery and development: review of select case studies including assay development considerations. Biomed Chromatogr 20:383–414
Acknowledgments
The authors thank the support of Fundacão para a Ciência e a Tecnologia (FCT, Portugal) through the fellowship SFHR/BD/84936/2012, involving the POPH (Programa Operacional Potencial Humano) which is cofounded by FSE (Fundo Social Europeu), and through the strategic project Pest-OE/SAU/UI0709/2014.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared no conflict of interest.
Rights and permissions
About this article

Cite this article
Ferreira, A., Rodrigues, M., Falcão, A. et al. HPLC–DAD Method for the Quantification of Carbamazepine, Oxcarbazepine and their Active Metabolites in HepaRG Cell Culture Samples. Chromatographia 79, 581–590 (2016). https://doi.org/10.1007/s10337-016-3063-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-016-3063-7