
Abstract
In drug discovery and development, it is very important to investigate the plasma protein binding (PPB) of a drug to better understand its in vivo fate. In this study, a rapid and low-cost solid-phase extraction (SPE) method was developed for determining the PPB. With this method, the total protein recovery of a blank human plasma sample was 83.7 %. The unbound drug was easily adsorbed by an ODS C18 SPE column, and the recovery of three known drugs was more than 90 %. Their PPBs obtained by the SPE were identical to the value reported by conventional techniques. In addition, more than 90 % of 4-amino-2-trifluoromethyl-phenyl retinate (ATPR), which is a novel all-trans retinoic acid derivative (ATRA), was bound to human plasma protein as determined by SPE, and this value was comparable with that obtained by our previously described gel filtration-based method. Considering its versatility, speed of separation, and low cost, SPE is a rapid and economical method for measuring PPB.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
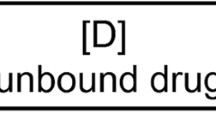
When circulating in the bloodstream, drugs bind to plasma proteins in varying degrees. Free and bound drug molecules undergo transport but only the free drug molecules are able to cross membrane barriers unless there is a specific transport system [1]. Then, the drug that crosses membrane barriers is distributed to tissues to undergo metabolism and glomerular filtration. Drug binding to plasma proteins is a reversible process, and, assuming there is only one reversible-binding site on the protein for a particular drug molecule, the binding between the drug and the protein can be described as follows:
where [D], [P], and [DP] are the free drug, free protein, and drug-protein complex concentrations, respectively, and k1 and k2 are the association and dissociation rate constants.
At equilibrium, the association constant (K) can be defined as follows:
According to the above equation, the bigger the K, the fewer free drug molecules are able to cross membrane barriers. Therefore, it is very important to investigate drug-protein interactions to fully understand the in vivo fate of a drug.
Many approaches have been used to investigate drug-protein binding. These methods can be divided into two groups, separative and non-separative methods [1]. A separative method is usually used to study drug-protein binding if the free drug concentration can be determined easily. Separative methods include equilibrium dialysis (ED), ultrafiltration (UF), parallel artificial membrane permeability assay (PAMPA), size-exclusion chromatography (SEC), capillary electrophoresis (CE), surface plasmon resonance-based assays (SPR) [1], microdialysis [2], and solid-phase microextraction (SPME) [3]. Some of these methods, such as CE, SPR, and microdialysis, are not widely used because they require expensive equipment. As for PAMPA, it is difficult to choose an appropriate membrane for different drugs, especially for batch analysis. In addition, due to its low column efficiency and poor protein recovery, SEC is rarely used nowadays [4]. Although ED and UF are suitable methods for drug-protein binding analysis because of their low cost and ease of use, nonspecific drug adsorption to the dialysis membrane or filter membrane can often occur [4, 5]. In addition, the two analysis methods are very slow.
In this study, we report a novel solid phase extraction (SPE) method to analyze drug-protein binding. SPE is a powerful and fast technique allowing rapid sample preparation.
SPE involves a partitioning of compounds between two phases, the mobile phase (pH 7.4 PBS or MeOH in this study) and stationary phase. For drug-protein binding analysis, the free drug is retained on the stationary phase, while that bound drug is eluted by pH 7.4 PBS. SPE has a number of advantages. For example, SPE reduces the analysis time if automated methods are used, and it needs only a small volume of sample (50–100 μL) and solvent. Nowadays there are many automatic SPE instruments on the market, and some of these commercial systems have automated SPE steps so that a number of cartridges can be used simultaneously (usually between 10 and 24) [6]. Furthermore, there are different stationary phases suitable for different compounds, and the stationary phases are classified as non-polar (e.g., C18), polar (e.g. NH2), ionic or mixed mode (C8/cation exchange). The versatility of SPE allows the use of this technique for many different drugs [7, 8].
In this study, we used ODS C18-filled SPE as an example to determine the binding constants of three known drugs and a novel drug (see Fig. 1 for the chemical structures of the four drugs).

Chemical structures of the four drugs. a All-trans retinoic acid (ATRA); b nimodipine; c 4-amino-2-trifluoromethyl-phenyl (ATPR); d phenacetin
Materials and Methods
Materials
All-trans retinoic acid (ATRA 98 %) is a commercial product from Beililai Biochemical Co., Ltd. (Beijing, China). Nimodipine (98 %) and phenacetin (98 %) were purchased from Sigma-Aldrich (Shanghai, China). 4-Amino-2-trifluoromethyl-phenyl retinate (ATPR) was synthesized from ATRA as an initial reagent as described in a previous report [9], and its purity was 99.73 % as confirmed by HPLC. A Bicinchoninic acid (BCA) Protein Assay Kit was purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Cleanert C18-SPE cartridges (100 mg/mL) were purchased from Agela Technologies Co., Ltd. (Tianjin, China). Fresh heparinized human plasma was obtained from the Blood Transfusion Centre of the First Affiliated Hospital of Anhui Medical University.
Preparation of Standard Solutions
ATRA, ATPR, nimodipine, and phenacetin were dissolved in methanol to give concentrations of 10 mg/mL. These stock solutions were protected from light and kept at −20 °C until used and then mixed with PBS pH 7.4 (0.05 M) to prepare standard solutions just prior to use.
Preparation of Plasma Samples
Human plasma was vortex mixed with ATRA, ATPR, nimodipine, and phenacetin for 3 min at room temperature at three concentration levels, i.e., 1, 5, and 50 mg/L for ATRA and ATPR; 0.5, 1.5, and 5 mg/L for nimodipine; 1, 5, and 10 mg/L for phenacetin.
Chromatographic Conditions
Analysis was carried out on an HPLC instrument with an SPD-20AV UV detector (Shimadzu LC-20AT system, Japan) equipped with a Hypersil ODS2 column (5 µm particle size, 250 mm × 4.6 mm).
The mobile phase for gradient elution of ATRA and ATPR consisted of methanol, acetonitrile, and 50 mmol/L acetate buffer (pH 4.5) with a volume ratio of 25:60:15 for the first 9 min, followed by 40:60:0 for 5 min and finally 25:60:15 for 8 min. The flow rate was 1.0 mL/min, and the detection wavelength was 345 nm for ATRA and 367 nm for ATPR (Shimadzu SPD-20AV UV detector, Japan). The injection volume was set at 20 μL. The drug concentrations in the mobile phase after eluting the samples with PBS were estimated using a pre-constructed standard calibration curve (y = 190.6 x + 961.4, where y is the integrated area, and x is the concentration of ATRA) with a range of 40–25,000 ng/mL and a correlation coefficient of 0.9998. The LOD was 15 ng/mL, and the LOQ was 40 ng/mL. For ATPR, over the concentration range 40–20,000 ng/mL, y = 141.9 x + 47.9, the correlation coefficient was 0.9997, and the LOD and LOQ were 15 and 40 ng/mL, respectively.
The mobile phase for nimodipine consisted of a mixture of MeOH-deionized water (60:40, v/v) and was delivered at a flow rate of 1.0 mL/min. Eluted peaks were detected at 237 nm. The drug concentrations in the mobile phase after eluting the sample with PBS were estimated using a pre-constructed standard calibration curve (y = 1.48 × 105 x + 3.018 × 103, where y is the integrated area, and x is the concentration of nimodipine) over the range 0.25–50 μg/mL. The correlation coefficient was 0.9994, and the LOD and LOQ were 0.08 and 0.25 μg/mL, respectively.
The mobile phase for phenacetin consisted of a mixture of MeOH-deionized water (20:80, v/v) and was delivered at a flow rate of 1.0 mL/min. Eluted peaks were detected at 249 nm. The drug concentrations in mobile phase after eluting the sample with methanol were estimated using a pre-constructed standard calibration curve (y = 9.18 × 104 x + 9.86 × 103, where y is the integrated area, and x is the concentration of phenacetin) over the range 0.20–50 μg/mL. The correlation coefficient was 0.9991, and the LOD and LOQ were 0.05 and 0.20 μg/mL, respectively.
Recovery of Total Protein by SPE
The SPE cartridges were activated with 2 mL MeOH, conditioned with 1 mL PBS pH 7.4 (0.05 M), and then they were loaded with 50, 100, and 250 μL blank human plasma. The analytes were eluted by washing the cartridge with pH 7.4 PBS (0.05 M), and 200 μL fractions of every elution were collected. The absorbance at 280 nm (E280) was used to obtain the elution curve of total protein [10, 11]. The concentration of total protein was determined using a bicinchoninic acid (BCA) protein assay kit to calculate the recovery of total protein. The standard calibration curve was y = 6.78 × 10−4 x + 0.13, where y is the integrated area, and x is the concentration of protein over the range 50–2000 μg/mL with a correlation coefficient of 0.9970. The LOD and LOQ were 5 and 50 μg/mL, respectively.
Recovery of Drug by SPE
The SPE cartridges were activated with 2 mL MeOH, conditioned with 1 mL PBS pH 7.4 (0.05 M), and then loaded with 50 μL of the standard solutions. The SPE cartridges were washed with 1 mL pH 7.4 PBS 0.05 M. Then the loaded drug was eluted using 1 mL methanol. The drug concentrations in MeOH were determined by HPLC as described in “Chromatographic Conditions” for plotting the elution curve and calculating the recovery of total drug.
Degree of PPB
The SPE cartridges were activated with 2 mL MeOH, conditioned with 1 mL PBS pH 7.4 (0.05 M), and then loaded with 50 μL of the drug-containing plasma samples. The SPE cartridges were washed with 1 mL pH 7.4 PBS 0.05 M to remove the protein-bound drug. Then the loaded drug was eluted using 1 mL methanol, and the drug concentrations in pH 7.4 PBS or MeOH were determined by HPLC as described in “Chromatographic Conditions”.
Results and Discussion
Recovery of Total Protein by SPE
The elution curves of blank human plasma samples are shown in Fig. 2. As can be seen from Fig. 2, the 50 and 100 μL blank human plasma samples were thoroughly washed with about 800 μL pH 7.4 PBS, but it needed at least 1400 μL pH 7.4 PBS when the plasma volume increased to 250 μL. In this study, the volume of all the samples for analysis was set at 50 μL, and the wash volume of PBS was 1000 μL. The total protein recovery for a 50-μL blank human plasma sample was 83.7 ± 2.8 % as analyzed by the BCA protein assay kit. These results indicated that the protein is able to pass through the SPE cartridges, and human plasma can be washed from these SPE cartridges. This can be explained as follows: according to the instructions for the SPE cartridges, the SPE packing material is spherical, and the average pore diameter and average particle size of the SPE packing material are 6 nm and 45 μm, respectively. As we know, α2-macroglobulin is the largest major nonimmunoglobulin protein in plasma, and its radius is about 9 nm [12]. This means that some protein is not able to pass through the internal pores of the SPE adsorbent. However, it can pass easily through the gaps of the spherical SPE adsorbent because the gap formed by the 45-μm ball particle could fill at least a 6.93-μm ball particle (If an octahedron is formed by close packing of equal spheres of radius r, then the radius of the sphere that can fit into the octahedral void is 0.154 r.)

Elution curve for a blank human plasma sample (n = 3)
The total protein recovery for the SPE method was less than 100 % (actually, 83.7 % in this study), but we also think that it can be used to measure PPB, like size-exclusion chromatography, which is used to measure PPB [1], and we previouly reported that the total protein recovery was 86 % using gel filtration [13]. This may due to the drug binding mainly to the protein that was eluted by the polar solvent. In other words, the bound drug will be eluted with the protein, and the free drug will be adsorbed by the SPE adsorbent. So, even if the total protein recovery is low, it can be separated effectively by SPE.
The sample separation speed is important for measuring plasma protein binding. This is because using SPE the binding equilbrium may be altered during measuring processes, e.g., by ultrafiltration, ultracentrifugation, and size-exclusion chromatography [4]. The more times it is used, the more the bound drug will be dissociated during the measuring process. Although ED possesses the advantage that equilibrium is maintained during the whole experiment, the equilibration times are long (about 12–48 h) [14]. In this study, we analyzed the plasma samples at 1.0 mL/min; at this flow speed, it needs only 2 min to complete the elution process. Considering its faster separation, the SPE may largely avoid drug-protein dissociation during the elution process.
Determination of the PPB Value for Nimodipine by SPE
The SPE cartridges were washed with 1 mL pH 7.4 PBS 0.05 M to remove the protein-bound drug. Then the loaded nimodipine (0.5, 1.5, and 5 μg/mL) was eluted using 1 mL methanol. Fractions of 200 μL from every elution of methanol were collected. The elution curves are shown in Fig. 3, and the recovery and PPB of nimodipine are listed in Table 1.

Elution curves for nimodipine standard solutions (n = 3)
Figure 3 and the recovery of nimodipine in Table 1 show that nimodipine was easily eluted from the SPE cartridges. The 0.5, 1.5, and 5 μg/mL nimodipine standard solutions were completely eluted using about 600 μL methanol. In this study, the elution volume of methanol for the nimodipine plasma samples was set at 1000 μL, and the recovery of nimodipine was more than 90 % (see Table 1). In addition, no nimodipine was found in the PBS eluate. This means that the unbound (free) nimodipine in the plasma samples was absorbed by the stationary phase of the SPE cartridges, and the protein-bound nimodipine could be easily eluted by the mobile phase.
As we can see from Table 1, the PPB of nimodipine was more than 96 % as determined by SPE, which is identical to the value of 95 % in the literature [15]. However, as we know, nimodipine is a water-insoluble drug, its pKa is 5.41 [15], and it will be neutral at physiological pH (7.4). Nimodipine is only one example of many insoluble and neutral drugs. Therefore, SPE is an ideal PPB analytical method for nimodipine and may even be suitable for many other insoluble and neutral drugs.
Determination of the PPB Value for Phenacetin by SPE
The SPE cartridges were washed with 1 mL pH 7.4 PBS 0.05 M to remove the protein-bound drug. Then the loaded phenacetin (1, 5, and 10 μg/mL) was eluted using 1 mL methanol. Fractions of 200 μL from every elution of methanol were collected. The elution curves are shown in Fig. 4, and the recovery and PPB of phenacetin are listed in Table 1.

Elution curves for phenacetin standard solutions (n = 3)
As can be seen from Fig. 4, the 1, 5, and 10 μg/mL phenacetin standard solutions were completely eluted with more than 400 μL methanol. In this study, the elution volume of methanol was 1000 μL for complete recovery of phenacetin. The recovery of phenacetin was more than 90 %, as shown in Table 1, and the values were slightly higher than those of nimodipine. This is due to the higher solubility of phenacetin, which means that it was eluted more easily with methanol.
As shown in Table 1, the PPB of phenacetin was about 30 %, which is near to the value of 30 % reported by Li et al. [16]. Furthermore, phenacetin is a water-soluble drug, and its pKa is 14.98 [17]. This study indicated that SPE is suitable for determining the PPB of phenacetin and may even be suitable for other water-soluble, non-dissociated drugs such as phenacetin.
Determination of the PPB Value for ATRA by SPE
As can be seen from Fig. 5, ATRA standard solutions at three different concentrations were adsorbed by SPE and then were eluted from SPE cartridges using 1 mL methanol. At 1, 5, and 50 μg/mL, the ATRA samples were almost completely eluted with 400 μL methanol. In this study, the volume of the mobile phase was set at 1000 μL. Table 1 shows that the recovery of ATRA was more than 95 %, and the PPB of ATRA determined by SPE was more than 90 %, which is close to the value of 93.3, 94.9, and 97.4 % at 1, 5, and 50 μg/mL, respectively [13].

Elution curves for ATRA standard solutions (n = 3)
ATRA is an acidic and poorly water-soluble drug widely used in the treatment of leukemia. In our pre-experiment (data not shown), ATRA was easily adsorbed by the dialysis membrane and filter membrane; thus, it is not really suitable to determine the PPB of ATRA by equilibrium dialysis and ultrafiltration. In this study, the protein-bound ATRA was found in the PBS solution after washing the sample with PBS. The unbound ATRA was easily adsorbed by the SPE adsorbent and then eluted with methanol and dissolved in methanol. This study indicated that SPE is suitable for determining the PPB of ATRA and may be suitable for many other poorly water-soluble and acidic drugs.
Determination of the PPB Value for ATPR by SPE
ATPR, a novel all-trans retinoic acid derivative, is a weakly basic drug with a lower solubility than ATRA. Considering that ATPR plays a major role in many biological processes, such as the superior ATPR differentiation-inducing activity involving, for example, SGC-7901, BEL-7402, HT-29, and MDA-MB-231 cell lines [18–20], it is important to understand the interaction of this compound with major carrier proteins.
As can be seen from Fig. 6, at 1 and 5 μg/mL, ATPR was almost completely eluted with 400 μL methanol, while, at 50 μg/mL, ATPR was eluted completely with 600 μL methanol. It seems that ATPR was adsorbed more strongly by SPE than ATRA. This may be due to the fact that ATPR is basic (the pka was 10.99, calculated by ChemAxon 15.3.9) and most likely fully protonated (positively charged) at pH 7.4, and it interacted with the weakly acidic free silanol groups on the SPE sorbent, while the weakly acidic ATRA did not participate in ionic interactions with negatively charged silanol groups.

Elution curves of ATPR standard solutions (n = 3)
ATPR is less polar than ATRA, and the recovery and PPB of ATPR are listed in Table 1. The high recovery of ATPR indicated that ATPR is not adsorbed irreversibly and can be eluted effectively. The PPB of ATPR determined by SPE was more than 90 %, which is close to the previously reported values of 90.5, 94.1, and 90.9 % at 1, 5, and 50 μg/mL, respectively. Compared with the previous study determining the PPB of ATPR by gel filtration (Sephadex LH-20), SPE was a more economical and faster method than gel filtration [13].
Conclusion
In drug discovery and development, it is very important to investigate PPB to fully understand the in vivo fate of a drug. There are many methods to determine PPB, but each one has its own advantages and disadvantages. Some need expensive equipment, some are time-consuming, and some have only a narrow range of applications. In this study, we confirmed that SPE could be used to determine the PPB of acidic or basic, poorly water-soluble or water-soluble drugs. SPE is a cheaper and faster method, and, of course, there are many different kinds of SPE suitable for a wide range of drugs.
References
Vuignier K, Schappler J, Veuthey JL, Carrupt PA, Martel S (2010) Drug-protein binding: a critical review of analytical tools. Anal Bioanal Chem 398:53–66
Shaw LH, Tsai TH (2012) Simultaneous determination and pharmacokinetics of protein unbound aspirin and salicylic acid in rat blood and brain by microdialysis: an application to herbal-drug interaction. J Chromatogr B 895–896:31–38
Vuckovic D, Zhang X, Cudjoe E, Pawliszyn J (2010) Solid-phase microextraction in bioanalysis: new devices and directions. J Chromatogr A 1217:4041–4060
Oravcova J, Bohs B, Lindner W (1996) Drug-protein binding sites new trends in analytical and experimental methodology. J Chromatogr B Biomed Appl 677:1–28
Howard ML, Hill JJ, Galluppi GR, Mclean MA (2010) Plasma protein binding in drug discovery and development. Comb Chem HighThroughput Screen 13:1–18
Berrueta LA, Gallo B, Vicente F (1995) A review of solid phase extraction: basic principles and new developments. Chromatographia 40:474–483
Huck CW, Bonn GK (2000) Recent developments in polymer-based sorbents for solid-phase extraction. J Chromatogr A 885:51–72
Żwir-Ferenc A, Biziuk M (2006) Solid Phase extraction technique-trends, opportunities and applications. Pol J Environ Stud 15:677–690
Shen J, Shi JB, Chen FH, Wang Y, Ruan JJ, Huang Y (2009) Synthesis and antitumor activity of all-trans retinoic acid derivatives. Chin Chem Lett 20:809–811
Bartley IM, Hodgson B, Walker JS, Holme G (1972) The use of acid alumina and sephadex LH-20 for the separation and characterization of ethanol-soluble peptides produced by bacillus brevis. Biochem J 127:489–502
Smith JE, Milch PO, Muto Y, Goodman DS (1973) The plasma transport and metabolism of retinoic acid in the rat. Biochem J 132:821–827
Pochon F, Amand B, Lavalette D (1978) Rotational relaxation of free and protease-bound alpha2-macroglobulin. J Biol Chem 253:7496–7499
Tang JH, Yao J, Waddad AY, Zhou JP, Chen FH (2013) Interaction of a novel all-trans retinoic acid derivative with bovine serum albumin and human plasma protein studied by gel filtration (sephadex LH-20) and fluorescence quenching method. Lat Am J Pharm 32:392–399
Vuignier K, Veuthey JL, Carrupt PA, Schappler J (2013) Global analytical strategy to measure drug-plasma protein interactions: from high-throughput to in-depth analysis. Drug Discovery Today. 18(21–22):1030–1034
Drugbank: http://www.drugbank.ca/drugs/DB00393
Li YF, Zhang XQ, Hu WY, Liu PX, Zhang ZQ (2013) Rapid screening of drug-protein binding using high-performance affinity chromatography with columns. J Anall Methods Chem 2013:1–7
Drugbank: http://www.drugbank.ca/drugs/DB03783
Hong FQ, Chen FH, Wu F, Chen HH (2011) Effects of 4-amino-2-trifluoromethyl-phenyl retinate on differentiation of human digestive system tumor cells in vitro. Cancer Res Prev Treat 38:1375–1379 (in Chinese with English abstract)
Wang B, Yan YW, Zhou JL, Zhou Q, Gui SY, Wang Y (2013) A novel all-trans retinoid acid derivatives inhibits the migration of breast cancer cell lines MDA-MB-231 via myosin light chain kinase involving p38-MAPK pathway. Biomed Pharmacother 67:357–362
Wang N, Ge JF, Pan CX, Peng XQ, Chen HH, Wang XQ, Tang J, Hu W, Chen FH (2013) Anti-tumor effect of 4-Amino-2-Trifluoromethyl-Phen-yl Retinate on human breast cancer MCF-7 cells via up-regulation of retinoid receptor-inducedgene-1. Biomed Pharmacother 67:687–692
Acknowledgements
This study was supported by the National Natural Science Foundation of China (no. 81274100), Natural Science Foundation of Anhui Province of China (no. 1408085QH189), and Key Project for the Excellent Higher Education of Anhui Province of China (no. 2013SQRL019ZD). Financial support was also provided by the Young and Middle-aged Academic Backbone from Anhui Medical University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Tang, J., Song, J., Liu, X. et al. Development of a Rapid and Low Cost Method for Measuring Plasma Protein Binding. Chromatographia 78, 1169–1174 (2015). https://doi.org/10.1007/s10337-015-2929-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-015-2929-4