
Abstract
Objectives
In this study, a novel targeted MRI contrast agent was developed by coating gadolinium oxide nanoparticles (Gd2O3 NPs) with β-cyclodextrin (CD)-based polyester and targeted by folic acid (FA).
Materials and methods
The developed Gd2O3@PCD–FA MRI contrast agent was characterized and evaluated in relaxivity, in vitro cell targeting, cell toxicity, blood compatibility and in vivo tumor MR contrast enhancement.
Results
In vitro cytotoxicity and hemolysis assays revealed that Gd2O3@PCD–FA NPs have no significant cytotoxicity after 24 and 48 h against normal human breast cell line (MCF-10A) at concentration of up to 50 µg Gd+3/mL and have high blood compatibility at concentration of up to 500 µg Gd+3/mL. In vitro MR imaging experiments showed that Gd2O3@PCD–FA NPs enable targeted contrast T1- and T2-weighted MR imaging of M109 as overexpressing folate receptor cells. Besides, the in vivo analysis indicated that the maximum contrast-to-noise ratio (CNR) of tumor in mice increased after injection of Gd2O3@PCD–FA up to 5.89 ± 1.3 within 1 h under T1-weighted imaging mode and reduced to 1.45 ± 0.44 after 12 h. While CNR increased up to maximum value of 1.98 ± 0.28 after injection of Gd2O3@PCD within 6 h and reduced to 1.12 ± 0.13 within 12 h.
Conclusion
The results indicate the potential of Gd2O3@PCD–FA to serve as a novel targeted nano-contrast agent in MRI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Magnetic resonance imaging (MRI) is one of the powerful modalities for noninvasive visualization and quantification of biological processes within the living organisms. Distinguishing disease tissue from normal, MRI provides important graphical images of the human body through the principles of nuclear magnetic resonance. Furthermore, excellent image resolution, specific morphological and functional imaging, as well as the absence of ionization radiation, make MRI a unique and popular technique compared to others [1, 2]. However, the low sensitivity of MRI is the main drawback of this technique causing poor contrast between different tissues. This problem originates from the fact that the difference between the numbers of atoms in the high- and low-energy states is negligible; so, compared with other imaging modalities, this is one of the drawbacks of MRI application [3]. To improve visibility and sensitivity of tissue and overcome low sensitivity of diagnostic examinations, a number of exogenous contrast agents (CAs) have been so far developed and applied in MRI studies [4]. The most widely clinically applied MRI contrast agents are gadolinium (Gd)-based compounds producing a positive contrast which mainly consist of central Gd3+ ion complexes with chelating ligand [5]. Notwithstanding all advantages of commonly applied Gd3+ complexes, they suffer from the release of gadolinium ion causing serious adverse effects such as nephrogenic systemic fibrosis and long-term deposition in brain tissue which forced food and drug administration to limit the use of such agents [6]. Nanotechnology bestowed stability, selectivity and biocompatibility on Gd-based CAs by small size, high surface area, easy labeling, stable coating and subsequently high bioavailability [7]. The size and surface properties of the nano-CAs could be easily controlled to specifically deliver high payloads of the active contrast agents to the target tissue and generate higher image contrast than that of commercially available CAs without leakage of toxic Gd ions [8]. Several studies have been rapidly expanding on Gd-containing nanoparticles (NPs) as effective contrast agents in preclinical and clinical applications [9, 10].
Various polymer coatings and inorganic materials have been investigated for surface modification of the Gd2O3 NPs to control leakage of Gd ions and enhance blood compatibility of the system resulting in high circulation time and sufficient targeting without toxicity [11,12,13,14]. So, the choice of suitable coating agent as surface modifier is one of the most important steps in the designing of Gd-based CAs. β-Cyclodextrin (CD) is a water-soluble cyclic oligosaccharide which contains a hydrophilic outer surface and a lipophilic central cavity and has gained interest due to its attractive cavity size, efficient drug loading, availability, biocompatibility and relatively low expense [15, 16]. These valuable features make CD a perfect coating agent to be applied in NP modification [17].
One of the most important aspects in the use of nanoparticles as contrast agent is the opportunity to target a desired area. Active targeting can efficiently promote NP internalization via receptor-mediated endocytosis and increase the efficacy of the payloads. Targeted contrast agents can improve tissue discrimination and provide more information about specific tissues [18, 19]. The ligand-mediated targeting depends on the affinity of the ligands on the surface of the nano-CAs leading to improve cellular uptake by the targeted receptors that are overexpressed on the diseased tissues or cells [20]. A variety of specific receptors has been identified as markers for targeting nanotherapeutics to cancer tissues, cells or even specific organelles within them. Among several targeting agents, folic acid (FA) as a water-soluble vitamin has attracted a great deal of attention due to its easy conjugation and high affinity to the large number of cancer cells bearing folate receptors [21, 22].
In the light of the above information, herein gadolinium oxide NPs were coated with FA-conjugated CD-based polyester (PCD). Although various FA-functionalized Gd2O3 have been introduced as a targeted CA [23,24,25], this is the first report in which FA–PCD conjugate was applied for Gd2O3 decoration to produce targeted biocompatible CA which could be employed for targeted cancer imaging. Thanks to the high specific surface area achieved by gadolinium oxide NPs and hydrophilic character of CD-based polyester layer, the high accessibility of water molecule to the core of Gd2O3 and consequently acceptable relaxivity could be expected, which is essentially beneficial in enhancing MR imaging. Also, application of DTPA-bis-anhydride (DTPA-DA) as monomer in condensation polymerization endows the produced polyester with myriad acid and amide functional groups as selective gadolinium ion chelating agents preventing toxic gadolinium leakage [26] and providing proper functionality for further functionalization. Moreover, the hydrophobic cavity of the CD suggests that the designed NPs could be exploited for loading of appropriate hydrophobic cargo via inclusion complexation and could be also introduced as a promising system utilizable as theranostic.
Materials and methods
Materials
DTPA, bare Gd2O3 NPs (< 100 nm), folic acid (FA) and β-cyclodextrin (CD), and media recipes for MCF-10A and M109 cells were purchased from Sigma-Aldrich Co (USA). Gd-DOTA (Dotarem®) was obtained from Bayer Health Care Pharmaceuticals Inc. (Montville, NJ, USA). Other materials were purchased from Merck KGaA (Darmstadt, Germany). Breast normal (MCF-10A) and M109 (malignant tumors of the mouse pulmonary system) cell lines were provided from National Cell Bank of Pasteur Institute, Iran (IPI).
Synthesis
Preparation of DTPA-bis-anhydride (DTPA-DA)
A modified procedure described in the literature was applied for the preparation of DTPA-bis-anhydride (DTPA-DA) [27,28,29]. Briefly, 0.125 mol of DTPA was mixed with 0.558 mol of dried acetic anhydride and 0.770 mol of dried pyridine. The resulting suspension was stirred for 24 h at 65 °C. Then the solid was filtered, washed with diethyl ether, and dried under vacuum.
Preparation of PCD-coated Gd 2 O 3 (Gd 2 O 3 /PCD)
Briefly, Gd2O3 (0.3 g) was initially dispersed in dried DMSO (10 mL). CD (0.1 mmol, 0.12 g) was then added to the as-prepared dispersion followed by the addition of 0.5 mL of dried triethylamine (TEA). The reaction was allowed to stir for 12 h to form a stable complex between hydroxyl group of CD and Gd2O3. In the next step, DTPA-DA (0.78 mmol, 0.28 g) was added to the mixture and stirred for another 12 h. Gd2O3/PCD was centrifuged (12,000 rpm) and washed three times with deionized water and ethanol, and finally dried in desiccator.
Conjugation of folic acid to Gd 2 O 3 /PCD (Gd 2 O 3 /PCD–FA)
Hydrazine derivative of FA (FA-NH2) was initially synthesized according to the previously reported method [30]. FA-NH2 was then conjugated on the surface of Gd2O3/PCD through EDC/NHS amidation to achieve targeted nano-CA (Gd2O3/PCD–FA). Briefly, 0.05 g of the prepared Gd2O3/PCD was dissolved in 10 mL DMSO. EDC (0.04 g) and NHS (0.03 g) were added to the mixture of Gd2O3/PCD in DMSO and stirred at room temperature for 24 h (500 rpm). Afterward, FA-NH2 (0.12 g) was added to the medium and stirred at room temperature for another 12 h. The reaction mixture was then centrifuged (8000 rpm) and the precipitate was washed three times with deionized water and ethanol, and dried in desiccator.
Characterization
Transmission electron microscope (TEM) images were obtained from a Hitachi H7650 transmission electron microscope with CCD imaging system at an acceleration voltage of 120 kV. Field emission scanning electron microscopy (FESEM) images were obtained on a FEI Magellan 400 microscope. X-ray diffraction (XRD) pattern was collected using X’Pert PRO MPDPANalytical (Netherland) X-ray diffractometer with Cu target (40 kV, 40 mA). The UV/Vis absorbance was measured with a Hewlett-Packard 8453 spectrometer (200–700 nm). FTIR spectra were collected on Tensor 27 (Bruker Cor., Germany) spectrometer using KBr technique (400–4000 cm−1 at room temperature). DLS analysis was achieved by Nicomp 380 ZLS Zeta potential/Particle sizer (PSS Nicomp, USA). Gd concentration was measured by inductively coupled plasma atomic emission spectroscopy (ICP-OES 730-ES, Varian). Thermogravimetric analyzes (TGA) were obtained by a thermogravimetric analyzer (METTLER TOLEDO). VSM measurements were done by VSM, 7400 model, Lakeshore Cryotronics Inc., OH, USA. Phantom, in vitro, and in vivo MR imaging were performed on a 3.0 T MRI scanner (Siemens Prisma MRI Scanner using head coil). A clinical Siemens Prisma MRI scanner was applied in this study which is considered as biosafety level 1 (BSL-1). All precautions related to BSL-1 were considered in this work [31].
Relaxivity measurements
To measure the longitudinal (T1) and transverse (T2) relaxation times on phantoms, Gd2O3@PCD–FA and Gd2O3 NPs were dispersed in water at various Gd concentrations (0, 0.04, 0.08, 0.16, 0.32, 0.64 and 1.28 mM) according to ICP results. Dotarem was prepared at the same Gd+3 concentrations as a control and all samples were dispersed in 2% agarose solution. The T1-weighted images were achieved using a conventional spin echo sequence with the following parameters: TR/TE = 50, 200, 400, 600, 800, 1100, 1300, 1500, 1800, 2000/11 ms, slice thickness = 5 mm, a flip angle of 90°, the number of signal averages of 3, 128 × 128 mm2 field of view, and 256 × 256 matrix dimensions. The T2-weighted images were acquired using a multi-spin echo (MSE) sequence with the same parameters except TR/TE = 3000/10, 30, 60, 90, 130, 170, 210, 240, 270, 350 ms, and slice thickness = 5 mm.
Cell culture
The MCF-10A (epithelial normal breast), M109 (malignant tumors of the mouse pulmonary system) and 4T1 (epithelial breast carcinoma) cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) and RPMI containing 10% fetal bovine serum (FBS), 100 units ML−1 penicillin and 100 μg mL−1 streptomycin. Cells were incubated at 37 °C with 5% CO2.
Cytotoxicity assay
In vitro cytotoxicity of Gd2O3@PCD–FA and Gd2O3 NPs were assessed against MCF-10A cells using MTT assay. Briefly, the cells were seeded into a 96-well plate (5 × 103 cells/well) and incubated at 37 °C under 5% CO2 atmosphere for 24 h. After that, the fresh medium containing NPs at different concentrations (0, 1.5, 3.12, 6.25, 12.5, 25, 50 µg mL−1) were added to the medium. After 24- and 48-h incubation, the media containing NPs were removed and 100 μL of MTT was added and incubated for another 4 h. Then the solution was removed, the cells were lysed, and the dye was dissolved in 100 μL of dimethyl sulfoxide. The plates were kept in the dark for 1 h before spectrophotometric determination. The amount of absorption in each well reflects the conversion of MTT to formazan by the metabolically viable cells and viability was calculated using an automated microplate reader at 570 nm [32].
Hemolysis assay
For hemolysis assay, the human red blood cells (HRBCs) were obtained according to the reported procedure [33]. Briefly, the fresh human blood was stabilized and treated with EDTA to remove the supernatant by centrifugation at 2000 rpm for 10 min and refined by successive rinsing with PBS buffer (pH 7.4). The suspension of HRBCs was diluted ten times with PBS buffer (pH 7.4), and then 200 mL of HRBCs suspension was added to 800 mL of each sample with different concentrations (1.95–1000 µg mL−1). Triton X-100 (800 mL, 2% v/v) and PBS buffer (800 mL, pH 7.4) were utilized as positive and negative controls, respectively. All samples were incubated at room temperature for 2 h. Finally, the samples were centrifuged at 10,000 rpm for 2 min, and the absorbance of the supernatant was measured by UV–visible spectrophotometer at 541 nm. The hemolytic activity percentages of the different samples were calculated by Eq. 1:
In vitro and in vivo MRI assay
M109 cells (5 × 106) were incubated with Gd2O3@PCD–FA and Gd2O3@PCD (without FA) NPs to study the targeting ability of NPs with different concentrations (0, 12.5 and 50 µg/mL) for 6 h in 37 °C. For comparison, 4T1 cells with negligible FA receptor expression (5 × 106) were incubated with NPs with the same NPs and concentrations under similar conditions. After incubation, the cells were washed with PBS buffer three times and resuspended in PBS buffer with a cell density of 1 × 106 cells/mL before MR imaging. All MR imaging measurements were performed with 3.0 T systems (3 T Siemens Prisma). T1-weighted MR images were acquired using a conventional spin-echo sequence under the following parameters: TR/TE =500/12 ms, 220 × 320 matrices, 82 × 120 mm field of view, 140 Hz/Px of bandwidth, a slice thickness of 3 mm. T2-weighted MR images using a fast spin-echo sequence was used to reduce acquisition time under the following parameters: TR/TE =3600/90 ms, 220 × 320 matrices, 82 × 120 mm field of view, 220 Hz/Px of bandwidth, and a slice thickness of 3 mm.
The animal tumor model was established on 4- to 5-week-old balb/c (25 g) mice by subcutaneously inoculating M109 cell lines (malignant tumors of the mouse pulmonary system). The mice were subjected to MR imaging studies 2 weeks after tumor inoculation when the tumor reached 1.0–2.0 cm in diameter. Mice were randomly divided into two groups (n = 5 mice/group). For in vivo MRI study, Gd2O3@PCD–FA NPs along with Gd2O3@PCD NPs (as a control group) were intravenously injected to a tail vein of anesthetized mice (0.1 mmol of Gd+3 per kg of mice body weight, 250 μL) in accordance with the literature (the mice were anesthetized by 10 and 100 mg/kg of xylazine and ketamine, respectively) [34, 35]. In vivo T1-weighted MR images were serially acquired before and also 30 min, 1, 6, and 12 h after injection.
The protocol of the in vivo study was performed in accordance with the European Community guidelines as accepted principles for the use of experimental animals and was approved by local ethical committee, Tehran University of Medical Sciences (TUMS), Tehran, Iran (Approval number: IR.TUMS.REC0.1394.1461). Signal intensity was calculated through drawing a ROI (region of interest) on tumor and background organs. Contrast-to-noise ratio (CNR) was calculated for regions of interest in the tumor area and background organs by Eq. (2):
where Senh and Sback are the mean signal intensity in the tumor region and background organ, respectively, and SDb is the standard deviation for background signal [36]. MRI scans for animals were obtained using the following protocols: for coronal T 1 -weighted MR images fast-sspin-echo protocol: TR: 600 ms; TE: 8.6 ms; FA: 150°; slice thickness: 2 mm; FOV: 110 mm; matrix size: 192 × 154 and NEX: 6. All MRI measurements were performed by a 3.0 Tesla Siemens Prisma MRI Scanner using the head coil.
Statistical analysis
Data were processed using Microsoft Excel 2013 software and presented as mean± standard error of the mean. Statistical analyzes were performed using ANOVA with fixed effect and independent t test. P values lower than 0.05 (p < 0.05) and more than 0.05 (p > 0.05) were considered as significant and insignificant differences, respectively.
Results and discussion
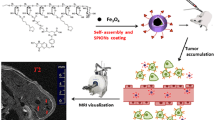
This study mainly focused on the preparation of targeted Gd2O3-based CA with the ability to distinguish cancerous cells from normal cells without Gd3+ ion leaching. The sequential steps for the preparation of the targeted nano-CA in MRI are shown in Fig. 1. Condensation polymerization was carried out between CD and DTPA-DA in the presence of Gd2O3 NPs resulting in CD-based polyester containing appropriate functional groups for chelating of Gd2O3 core and for further functionalization as well. Considering the availability, low cost, simple polymerization procedure and also proper affinity of DTPA towards Gd+3 ions, in this work, DTPA was selected as monomer to prepare cyclodextrin-based polymer as a coating agent. FA-NH2 was then conjugated to the newly appeared acidic groups on the surface of Gd2O3 NPs to equip the CA with active targeting agent.

Schematic representation of the synthesis of the targeted nano-CA in MRI and its application
Characterization
As shown in Fig. 2a, the FTIR spectrum of CD shows characteristic peaks at 3378 and 2926 cm−1, corresponding to the O–H and C-H stretching vibrations. Furthermore, the peaks at 1645 and 1029 cm−1 are related to bending vibration of O–H and stretching vibration of C–O–C glucose units of CD. The spectrum of DTPA-DA shows absorbtion peaks at 1822, 1776 and 1113 cm−1 which are attributed to the asymmetric and symmetric stretching vibration of C=O and C-N stretching in anhydride, respectively (Fig. 2a). The appeared bands in the spectrum of PCD (Fig. 2a) at around 1740 and 1634 cm−1 could be ascribed to the C=O asymmetric stretching vibration of ester groups and COOH starching in the form of carboxylate, respectively, indicating formation of PCD. The observed absorption frequencies in FTIR of pure Gd2O3 nanoparticles (Fig. 2b) include stretching vibrations of O–H at ~3415 cm−1, the Gd=O at ~1492 cm−1, Gd–O–Gd at ~1393 cm−1 and Gd-O ~ 540 cm−1 [37]. In the spectrum of Gd2O3@PCD, the absorption peaks at around 1739 and 1634 cm−1 are attributed to the C=O stretching vibrations of ester and carboxylate groups, respectively. Figure 2b shows the spectrum of the Gd2O3@PCD–FA NPs. The characteristic peaks of Gd2O3 and the polymer layer have been preserved. The peak at 1621 cm−1 which is attributed to the formation of amide bands after Gd2O3@PCD modification with FA-NH2 confirms successful attachment of FA on the surface of the NPs [38].

FTIR spectra of (a) CD, DTPA-DA and PCD. b Gd2O3, Gd2O3@PCD and Gd2O3@PCD–FA NPs
The amount of FA conjugation with Gd2O3@PCD was calculated by UV–Vis spectroscopy using different FA concentrations (0.14, 0.18, 0.23, and 0.28 mg/mL) in the supernatant by measuring the difference between the initially added FA and FA in the supernatant (λmax = 447 nm, r2 = 0.9991 with excellent linearity). The experiments were conducted three times. The amount of FA loaded on the surface of NPs was about 6%. Figure 3a shows the UV–Vis spectra of Gd2O3, Gd2O3@PCD and Gd2O3@PCD–FA, respectively. There was no significant difference between spectra of Gd2O3 and Gd2O3@PCD NPs, while peaks for folic acid at λ = 282 and 362 nm could be seen for Gd2O3@PCD–FA. Significant shift for the absorption peak of the FA in Gd2O3@PCD–FA confirmed the conjugation of FA to Gd2O3@PCD NPs [39, 40]. The Gd concentration on the final NPs was also estimated to be approximately 60% by inductively coupled plasma atomic emission spectroscopy (ICP-AES). The crystallography of the product was verified by powder X-ray diffraction (XRD) (Fig. 3b). It is clear that all the diffraction peaks of the Gd2O3@PCD–FA NPs are well matched with standard Gd2O3 and no other impurity peaks could be observed (Ref code no. 01–076-0155) [41, 42]. This pattern confirms the existence of Gd2O3 on the final NPs. The XRD pattern of Gd2O3@PCD–FA NPs showed a slight decrease in the intensity in comparison with Gd2O3 which could be due to the presence of PCD–FA on the surface of Gd2O3.

a UV–Vis spectra obtained at different steps of the NPs modification to confirm FA conjugation. b XRD patterns of Gd2O3@PCD–FA and Gd2O3 NPs
As shown in Fig. 4, all nanoparticles were in spherical shape and proper size distribution. Images of FESEM and TEM revealed that NPs were spherical and uniform in morphology and could be visualized separately with clear grains in nano-dimensions less than 100 nm (range 75–95 nm).

a FESEM micrographs and b TEM images of Gd2O3@PCD–FA
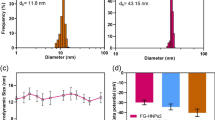
The size and polydispersity index (PDI) obtained by DLS showed that Gd2O3nanoparticles had a hydrodynamic diameter distribution of 96 ± 7.6 nm with a PDI of 0.36, while Gd2O3@PCD–FA NPs was 131 ± 4.6 nm with PDI of 0.38 (Table 1). However, despite their different sizes, PDIs of the NPs (as an index of the nanoparticle dispersion) showed acceptable ranges of less than 0.5. The results were in good agreement with the results of FESEM/TEM images of the NPs (Fig. 4). However, differences in the size of the NPs between DLS and FESEM/TEM measurements could be attributed to the hydrodynamic radius and water-swollen NPs in DLS measurement, whereas TEM/SEM represented an estimation of NP diameter in dried state [32]. Zeta potentials of Gd2O3 and Gd2O3@PCD–FA NPs were (+23.1 mv) and (-5.27 mv), respectively, that confirm Gd2O3 coating with PCD. The positive charge of the naked Gd2O3 NPs reduced to negative which is due to the negative charge of the polymer layer on the surface of the NPs after coating resulted from hydroxyl and carboxyl groups of the polymer.
Magnetic property measurements
The magnetic property of the NPs was evaluated by field-dependent magnetization measurements at 300 K. For paramagnetic materials, there is a linear relationship between magnetization (M) and applied field (H) with positive slope [43]. The saturated magnetization (Ms) of Gd2O3@PCD–FA NPs was found to be 0.63 emu/g (Fig. 5a), which is smaller than the value of the Gd2O3NPs (1emu/g) (Fig. 5b) due to the PCD layer that was in agreement with the literature [44].

a Magnetometry graphs for Gd2O3, b Gd2O3@PCD–FA NPs (magnetization (emu/g) plotted as a function of the applied field). c TGA curves of Gd2O3 and Gd2O3@PCD–FA NPs under a constant N2 flow
Thermogravimetric analysis (TGA)
TGA analysis under a constant N2 flow was performed for Gd2O3 and Gd2O3@PCD–FA NPs to achieve some structural information about the surface modification of the Gd2O3 NPs and determine the mass drop in weight (%) of the materials (Fig. 5c). The first step of the weight loss of about 4% in thermogram of Gd2O3 NPs can be seen over the range of about 100–270 °C indicating the evaporation of residual solvent and adsorbed water on the surface of the NPs. Comparison between Gd2O3 and Gd2O3@PCD–FA thermograms (Fig. 5c) indicated that the main weight loss of about 25% at the range 270–700 °C could be ascribed to the removal of PCD–FA shell on the surface of Gd2O3@PCD–FA NPs (Fig. 5c) [45, 46].
Relaxivity measurement
To evaluate the potential application of Gd2O3@PCD–FA NPs as a MR contrast agent, their magnetic relaxation properties was investigated compare to Gd2O3 and Dotarem. T1- and T2-weighted images were taken using aqueous suspensions of the NPs at certain concentrations. The Gd2O3@PCD–FA NPs and Gd2O3 were found to efficiently shorten the longitudinal relaxation time (T1) and significantly increase signal intensity in T1-weighted images compare to Dotarem (Fig. 6). However, the degree of the bright contrast enhancement in T1-weighted images for all groups was found to be directly related to the concentration of Gd ions. For the quantitative evaluation, the longitudinal (r1) and transverse (r2) relaxivities of the particles were calculated by measuring the relaxation rate as a function of Gd ion concentration (Table 2). The r1 values for Gd2O3@PCD–FA NPs, Gd2O3, and Dotarem were achieved to be 3.95, 4.86, and 2.79, respectively. According to other studies, the r1 of Gd2O3 is higher than standard approved Gd-based contrast agents [47, 48]. The partial reduction of r1 for Gd2O3@PCD–FA NPs compared to Gd2O3 could be due to the PCD as a shell which reduced the water molecule accessibility to the Gd2O3 NPs. The low r2/r1 ratio (1.16) showed that the T1-shortening effect was dominant over the T2 effect, suggesting the bright contrast enhancement in T1-weighted images. In the study of Fang et al., hydrophobicity and hydrophilicity of the polymer layer were evaluated. They observed reduced r1 and r2 values of Gd2O3–OA–CTAB nanoparticles (OA = oleic acid and CTAB = cetyltrimethylammonium bromide) compared to Gd2O3–PVP nanoparticles (PVP = polyvinyl pyrrolidone) [49]. It has been suggested that the polar C=O groups in the polymer coating layer on the surface of Gd2O3 endow the contrast agent with acceptable water accessibility. Appropriate relaxivity of Gd2O3@PCD–FA in our study even after coating with PCD as a hydrophilic polymer layer could be due to the presence of polar carbonyl groups on the surface of Gd2O3 core. The r1 and r2 values depend on various factors such as the type of NPs, particle and hydrodynamic diameter and the properties of ligands or coating agents surrounding the Gd+3 ions [49] and more studies need to be performed to find the optimum coating layer thickness and particle size of the target CA.

Longitudinal (a) and transverse (b) relaxivity plot of aqueous suspension of Dotarem, Gd2O3@PCD–FA and Gd2O3 NPs. c T1-weighted and d T2-weighted images of Gd2O3@PCD–FA, Gd2O3, and Dotarem in water
Cytotoxicity assay
Prior to the applications of Gd2O3@PCD–FA NPs for in vitro and in vivo MR imaging, the preliminary potential cytotoxicity of nanoparticles was evaluated via an MTT [3-(4,5-dimethylthiazol-2 yl)-2,5-diphenyltetrazolium bromide] assay on MCF-10A cell lines (human normal breast cell line). As shown in Fig. 7, no obvious decrease in cell viability was observed with various concentrations of Gd2O3@PCD–FA NPs (up to 50 µg/mL) for 24 h incubation time. Even at the concentration of 50 µg/mL, the cell viability still remained above 80%. Increasing incubation time of the NPs to 48 h, the cell survival rate decreases with a concentration-dependent trend and down to 79.4% within 50 µg/mL. Gd2O3 exhibited significant cytotoxicity within 24 and 48 h compared to Gd2O3@PCD–FA NPs that could be related to the leakage of Gd ions and toxicity of Gd ions (p < 0.005). It could be proposed that Gd2O3 coating by PCD led to reduction of Gd leakage, while Gd2O3@PCD–FA NPs has more free diffusion through phospholipid cell membrane than Gd2O3. The leakage of Gd+3 ions liberated from Gd2O3@PCD–FA was measured by ICP-AES. A solution of the sample containing 1.0 mM Gd in simulated physiological condition (i.e., pH 7.4, 0.1 mM glucose solution, at room temperature and 1 atm) was prepared and the concentration of Gd+3 ions in the supernatant was determined after 12 and 24 h. The results were under the detection limit of the ICP-AES (i.e., < 0.1 ppm Gd) showing the high affinity of the prepared polymer to the Gd+3 ions. These results indicated that Gd2O3@PCD–FA NPs could have little cytotoxicity at the given concentration range, which is essential to their further biological applications.

Cell viabilities for normal human breast cell line (MCF-10A) exposed to the NPs. a Gd2O3@PCD–FA and b Gd2O3 in 24 and 48 h incubation time at different Gd+3 concentrations. c Comparison of the cell viability between Gd2O3@PCD–FA and Gd2O3 NPs at 24 h incubation time. d Hemolytic activity and photographs of RBCs treated with Gd2O3@PCD–FA and Gd2O3 NPs at different Gd+3 concentrations
Hemolysis assay
Hemocompatibility of NPs could be confirmed by hemolysis assay to evaluate biosafety of NPs on erythrocytes [50]. When NPs are injected into the blood, detrimental interaction of the particles with blood constituents must be avoided. As shown in Fig. 7d, the percentage of hemolytic activity for the NPs in the studied concentration range (1.95–500 µg Gd+3/mL) were less than 6% (standard acceptance limit) suggesting their acceptable hemocompatibility [47]. The maximum hemolytic activity percentage of Gd2O3@PCD–FA NPs was 11.75% (concentration 1000 µg /mL). It was observed that the hemolytic percentage of the NPs depended on its concentrations.
A significant difference in hemolytic activity was observed between Gd2O3 and Gd2O3@PCD–FA. It could be due to toxicity of Gd ions which cause interaction between these particles and red blood cells, damaging most of red blood cells [51]. In the present study, PCD as coating layer led to blood compatibility of Gd2O3. The results also suggested that Gd2O3@PCD–FA NPs were suitable for a wide safety margin in blood-contacting applications and suitability for intravenous administration.
In vitro MRI measurements
To confirm the cancer cell targeting ability of the Gd2O3@PCD–FA NPs and MR imaging performance, both M109 cells with folate receptor overexpression and 4T1 cells with negligible FA receptor expression were incubated with the particles with different concentrations (0, 12.5 and 50 μg/mL) for 6 h. Both T1- and T2-weighted MR images and the MR signal intensity of the cells were collected. As shown in Fig. 8, the T1-weighted MR images showed a significant enhancement in M109 cells, while the T2-weighted MR images of the same cells showed a marked signal drop. A quantitative measure of the MR signal change further showed that when incubated with the Gd2O3@PCD–FA NPs (50 µg/mL) for 6 h, the T1 MR signal intensity of M109 cells was four times higher than the control cells without treatment, and the T2 MR signal intensity of the same cells was dropped 8 times lower than that of control cells.

a T1- and T2-weighted MR images of Gd2O3@PCD–FA and Gd2O3@PCD (without FA) NPs in M109 and 4T1 cells at different concentration of NPs after incubation for 6 h on 3T MR system. b Signal intensity analysis for T1-weighted MR images. c Signal intensity analysis for T2-weighted MR images
In contrast, under the same condition, the T1 MR signal intensity of 4T1 cells was only 1.9 times higher than that of the control 4T1 cells without treatment, and the T2 MR signal intensity was just dropped to 51% when compared with that of control 4T1 cells. The significant difference between M109 cells and 4T1 cells in MR imaging should be ascribed to the specific cellular uptake of the Gd2O3@PCD–FA NPs into M109 cells due to overexpressing folate receptor compared to 4T1 [52, 53]. To further prove that the specific binding and uptake of the particles is mediated through the specific interaction between FA and FRs that are overexpressed onto the cell surface, M109 cells were preincubated with NPs without FA (Gd2O3@PCD) and treated with the same concentrations for 6 h. T1- and T2-weighted MR imaging data showed that the T1 MR signal intensity of M109 cells was only about 2.3 times higher than that of the untreated control cells (50 µg/mL), and T2 MR signal intensity (50 µg/mL) was just dropped to 43% when compared with that of the control cells. This experiment clearly indicates that the specific binding and uptake of the particles should be mediated by the FA/FR interaction on the surface of cells. Taken together, our results suggest that the Gd2O3@PCD–FA NPs can specifically affect both T1 and T2 MR signals through FR-mediated cellular binding and uptake.
In vivo MR imaging
The targeted MR imaging of cancer cells using Gd2O3@PCD–FA NPs was further performed using an animal tumor model. Serial T1-weighted images were performed using fast-spin-echo sequence to measure signal enhancement at different time intervals after injection in tumor area (Fig. 9a, b). After intravenous injection of Gd2O3@PCD–FA, the contrast of the tumor position was gradually enhanced. To quantify the contrast, the tumor was identified as the region of interest and the contrast-to-noise ratio (CNR) values for each animal were determined. The CNR of tumor for Gd2O3@PCD–FA was increased from 1.00 ± 0.3 to 5.89 ± 1.3 within 1 h after injection and gradually reduced to 1.45 ± 0.44 within 12 h. To study the targeting ability of the NPs, Gd2O3@PCD as a control was injected with the same Gd+3 concentrations. The CNR for Gd2O3@PCD after injection was obtained as 1.98 ± 0.28 after 6 h, and reduced to 1.12 ± 0.13 after 12 h (Fig. 9c, p value < 0.05). As shown in Fig. 9a, the Gd2O3@PCD–FA NPs showed T1-weighted positive contrast-enhanced MR imaging capability of the tumor tissues after the intravenous administration. The results revealed that Gd2O3@PCD–FA NPs exhibited much higher contrast than Gd2O3@PCD throughout the imaging periods (p value < 0.05). The higher tumor accumulation of Gd2O3@PCD–FA NPs could clearly and selectively enhance the contrast at the tumor area in T1-weighted MR images, which could be attributed to the targeting ability of NPs due to the overexpression of FA receptors on the M109 cell surface [53].

T1-weighted images of mice tumor injected with (a) Gd2O3@PCD–FA NPs and b Gd2O3@PCD. c CNR analysis for T1-weighted images in tumor. Data represent mean ± SEM (n = 5), *p < 0.05, *p < 0.05, **p < 0.01
The gradual reduction of CNR for the Gd2O3@PCD–FA NPs was most likely due to their long circulation time and targeting ability [54,55,56]. The comparison of CNR values between Gd2O3@PCD and Gd2O3@PCD–FA confirmed the difference between EPR effect and the key role of FA as active targeting agent facilitating the cell penetration of the NPs. The use of Gd2O3@PCD–FA NPs with appropriate T1 relaxivity as contrast agent may significantly improve the sensitivity of T1 imaging, which is extremely important for accurate detection and early diagnosis of cancer.
Conclusion
In summary, we have successfully designed and synthesized a nanocomposite composed of gadolinium oxide NPs coated with polycyclodextrin (PCD) and targeted with folate with overall diameter of less than 100 nm as potential targeted MRI contrast agent. The in vitro toxicity studies demonstrated that PCD as a shell could significantly improve biosafety and excluded the toxicity risk of Gd2O3 as MRI contrast agent. The longitudinal relaxivity value (r1) of the Gd2O3@PCD–FA NPs was higher than that of the clinical contrast agent (Dotarem) in aqueous suspension. Although PCD partially reduced relaxivity of the NPs in aqueous suspension through reducing water molecule accessibility to Gd2O3, it could significantly improve cell viability. In vitro MRI measurements confirmed selective and enhanced accumulation of Gd2O3@PCD–FA intracellular uptake in M109 in cancer cells compared to that found in 4T1 cells due to folic acid as targeting ligands, and data confirm the important role of the targeting moiety. In vivo MRI studies demonstrated that Gd2O3@PCD–FA NPs had higher contrast enhancement in tumor area than Gd2O3@PCD. The different distribution of two contrast agents within 1 h post-injection in tumors showed that the Gd2O3@PCD–FA NPs could accumulate into tumor more than Gd2O3@PCD due to expression of FA receptors onto the cell surfaces. Furthermore, a higher circulation time for the CA to accumulate in tumors was observed which could be due to the nanonization as well as active targeting. The selective toxicity, acceptable blood compatibility of the Gd2O3@PCD–FA NPs compared to bare Gd2O3 and its higher relaxivity than commercial Dotarem suggest these targeted NPs as a promising targeted CA for early diagnosis of cancer via MRI. The presence of hydrophobic cavity of CD in structure of the target NPs endows them with the ability to load and deliver any hydrophobic cargos which make these NPs valuable to be considered as promising theranostic agents.
References
Lee SH, Kim BH, Na HB, Hyeon T (2014) Paramagnetic inorganic nanoparticles as T1 MRI contrast agents. Wiley Interdiscip Rev Nanomed Nanobiotechnol 6(2):196–209
Azizian G, Riyahi-Alam N, Haghgoo S, Moghimi HR, Zohdiaghdam R, Rafiei B, Gorji E (2012) Synthesis route and three different core-shell impacts on magnetic characterization of gadolinium oxide-based nanoparticles as new contrast agents for molecular magnetic resonance imaging. Nanoscale Res Lett 7(1):549–559
Tegafaw T, Xu W, Lee SH, Chae KS, Cha H, Chang Y, Lee GH (2016) Ligand-size and ligand-chain hydrophilicity effects on the relaxometric properties of ultrasmall Gd2O3 nanoparticles. AIP Adv 6(6):065114
Lee N, Hyeon T (2012) Designed synthesis of uniformly sized iron oxide nanoparticles for efficient magnetic resonance imaging contrast agents. Chem Soc Rev 41(7):2575–2589
Narmani A, Farhood B, Haghi-Aminjan H, Mortezazadeh T, Aliasgharzadeh A, Mohseni M, Najafi M (2018) Gadolinium nanoparticles as diagnostic and therapeutic agents: their delivery systems in magnetic resonance imaging and neutron capture therapy. J Drug Deliv Sci Technol 44:457–466
Abraham J, Thakral C, Skov L, Rossen K, Marckmann P (2008) Dermal inorganic gadolinium concentrations: evidence for in vivo transmetallation and long-term persistence in nephrogenic systemic fibrosis. Br J Dermatol 158(2):273–280
Challa R, Ahuja A, Ali J, Khar R (2005) Cyclodextrins in drug delivery: an updated review. AAPS Pharm Sci Tech 6(2):E329–E357
Waters EA, Wickline SA (2008) Contrast agents for MRI. Basic Res Cardiol 103(2):114–121
McDonald MA, Watkin KL (2003) Small particulate gadolinium oxide and gadolinium oxide albumin microspheres as multimodal contrast and therapeutic agents. Invest Radiol 38(6):305–310
Rahman AA, Vasilev K, Majewski P (2011) Ultra small Gd2O3 nanoparticles: absorption and emission properties. J Colloid Interface Sci 354(2):592–596
Cheung ENM, Alvares RD, Oakden W, Chaudhary R, Hill ML, Pichaandi J, Mo GC, Yip C, Macdonald PM, Stanisz GJ (2010) Polymer-stabilized lanthanide fluoride nanoparticle aggregates as contrast agents for magnetic resonance imaging and computed tomography. Chem Mater 22(16):4728–4739
Ahrén M, La Selegård, Klasson A, Söderlind F, Abrikossova N, Skoglund C, Tr Bengtsson, Engström M, Käll P-O, Uvdal K (2010) Synthesis and characterization of PEGylated Gd2O3 nanoparticles for MRI contrast enhancement. Langmuir 26(8):5753–5762
Zohdiaghdam R, Riyahi-Alam N, Moghimi H, Haghgoo S, Alinaghi A, Azizian G, Ghanaati H, Gorji E, Rafiei B (2013) Development of a novel lipidic nanoparticle probe using liposomal encapsulated Gd2O3–DEG for molecular MRI. J Microencapsul 30(7):613–623
Akai H, Shiraishi K, Yokoyama M, Yasaka K, Nojima M, Inoue Y, Abe O, Ohtomo K, Kiryu S (2018) PEG-poly (l-lysine)-based polymeric micelle MRI contrast agent: Feasibility study of a Gd-micelle contrast agent for MR lymphography. J Magn Reson Imaging 47(1):238–245
Trotta F, Zanetti M, Cavalli R (2012) Cyclodextrin-based nanosponges as drug carriers. Beilstein J Org Chem 8(1):2091–2099
Vyas A, Saraf S, Saraf S (2010) Encapsulation of cyclodextrin complexed simvastatin in chitosan nanocarriers: a novel technique for oral delivery. J Incl Phenom Macrocycl Chem 66(3–4):251–259
Zhou Q, Guo X, Chen T, Zhang Z, Shao S, Luo C, Li J, Zhou S (2011) Target-specific cellular uptake of folate-decorated biodegradable polymer micelles. J Phys Chem B 115(43):12662–12670
Hengerer A, Grimm J (2006) Molecular magnetic resonance imaging. Biomed Imaging Interv J 2(2):e8
Achilefu S (2004) Lighting up tumors with receptor-specific optical molecular probes. Technol Cancer Res Treat 3(4):393–409
Daryasari MP, Akhgar MR, Mamashli F, Bigdeli B, Khoobi M (2016) Chitosan-folate coated mesoporous silica nanoparticles as a smart and pH-sensitive system for curcumin delivery. RSC Adv 6(107):105578–105588
Chen C, Ke J, Zhou XE, Yi W, Brunzelle JS, Li J, Yong E-L, Xu HE, Melcher K (2013) Structural basis for molecular recognition of folic acid by folate receptors. Nature 500(7463):486
Conte C, Fotticchia I, Tirino P, Moret F, Pagano B, Gref R, Ungaro F, Reddi E, Giancola C, Quaglia F (2016) Cyclodextrin-assisted assembly of PEGylated polyester nanoparticles decorated with folate. Colloids Surf B Biointerfaces 141:148–157
Oyewumi MO, Yokel RA, Jay M, Coakley T, Mumper RJ (2004) Comparison of cell uptake, biodistribution and tumor retention of folate-coated and PEG-coated gadolinium nanoparticles in tumor-bearing mice. J Control Release 95(3):613–626
Nakamura T, Kawano K, Shiraishi K, Yokoyama M, Maitani Y (2014) Folate-targeted gadolinium-lipid-based nanoparticles as a bimodal contrast agent for tumor fluorescent and magnetic resonance imaging. Biol Pharm Bull 37(4):521–527
Shah SA, Khan MA, Arshad M, Awan S, Hashmi M, Ahmad N (2016) Doxorubicin-loaded photosensitive magnetic liposomes for multi-modal cancer therapy. Colloids Surf B Biointerfaces 148:157–164
Kalender W (2004) Classic papers in modern diagnostic radiology. Springer Science & Business Media.
Duarte M, Gil M, Peters J, Colet J, Elst LV, Muller R, Geraldes C (2001) Synthesis, characterization, and relaxivity of two linear Gd (DTPA)− polymer conjugates. Bioconjug Chem 12(2):170–177
Vahedi S, Tavakoli O, Khoobi M, Ansari A, Faramarzi MA (2017) Application of novel magnetic β-cyclodextrin-anhydride polymer nano-adsorbent in cationic dye removal from aqueous solution. J Taiwan Inst Chem Eng 80:452–463
Heydarnezhadi S, Alam NR, Haghgoo S, Ghanaati H, Khoobi M, Gorji E, Rafiei B, Nikfari B, Amirrashedi M (2016) Glycosylated Gadolinium as Potential Metabolic Contrast Agent vs Gd-DTPA for Metabolism of Tumor Tissue in Magnetic Resonance Imaging. Appl Magn Reson 47(4):375–385
Anbharasi V, Cao N, Feng SS (2010) Doxorubicin conjugated to D-α-tocopheryl polyethylene glycol succinate and folic acid as a prodrug for targeted chemotherapy. J Biomed Mater Res A 94(3):730–743
Richmond JY, McKinney RW (1993) Biosafety in microbiological and biomedical laboratories. DIANE Publishing.
Hou W, Xia F, Alfranca G, Yan H, Zhi X, Liu Y, Peng C, Zhang C, de la Fuente JM, Cui D (2017) Nanoparticles for multi-modality cancer diagnosis: simple protocol for self-assembly of gold nanoclusters mediated by gadolinium ions. Biomaterials 120:103–114
Akrami M, Khoobi M, Khalilvand-Sedagheh M, Haririan I, Bahador A, Faramarzi MA, Rezaei S, Javar HA, Salehi F, Ardestani SK (2015) Evaluation of multilayer coated magnetic nanoparticles as biocompatible curcumin delivery platforms for breast cancer treatment. RSC Adv 5(107):88096–88107
Ahmad MW, Xu W, Kim SJ, Baeck JS, Chang Y, Bae JE, Chae KS, Park JA, Kim TJ, Lee GH (2015) Potential dual imaging nanoparticle: Gd 2 O 3 nanoparticle. Sci Rep 5:8549
Kim CR, Baeck JS, Chang Y, Bae JE, Chae KS, Lee GH (2014) Ligand-size dependent water proton relaxivities in ultrasmall gadolinium oxide nanoparticles and in vivo T 1 MR images in a 1.5 T MR field. Phys Chem Chem Phys 16 (37):19866-19873.
Ghaghada KB, Ravoori M, Sabapathy D, Bankson J, Kundra V, Annapragada A (2009) New dual mode gadolinium nanoparticle contrast agent for magnetic resonance imaging. PLoS ONE 4(10):e7628
Ma J, La LTB, Zaman I, Meng Q, Luong L, Ogilvie D, Kuan HC (2011) Fabrication, structure and properties of epoxy/metal nanocomposites. Macromol Mater Eng 296(5):465–474
Yang X, Wang Y, Huang X, Ma Y, Huang Y, Yang R, Duan H, Chen Y (2011) Multi-functionalized graphene oxide based anticancer drug-carrier with dual-targeting function and pH-sensitivity. J Mater Chem 21(10):3448–3454
Huang P, Xu C, Lin J, Wang C, Wang X, Zhang C, Zhou X, Guo S, Cui D (2011) Folic acid-conjugated graphene oxide loaded with photosensitizers for targeting photodynamic therapy. Theranostics 1:240
Zhao F, Yin H, Zhang Z, Li J (2013) Folic acid modified cationic γ-cyclodextrin-oligoethylenimine star polymer with bioreducible disulfide linker for efficient targeted gene delivery. Biomacromol 14(2):476–484
Liu Y, Yang P, Wang W, Dong H, Lin J (2010) Fabrication and photoluminescence properties of hollow Gd 2 O 3: Ln (Ln = Eu3+, Sm3+) spheres via a sacrificial template method. Cryst Eng Comm 12(11):3717–3723
Kumar S, Meena VK, Hazari PP, Sharma RK (2016) FITC-Dextran entrapped and silica coated gadolinium oxide nanoparticles for synchronous optical and magnetic resonance imaging applications. Int J Pharm 506(1–2):242–252
Rohrer M, Bauer H, Mintorovitch J, Requardt M, Weinmann H-J (2005) Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths. Invest Radiol 40(11):715–724
Leng Y, Sato K, Shi Y, Li J-G, Ishigaki T, Yoshida T, Kamiya H (2009) Oxidation-resistant silica coating on gas-phase-reduced iron nanoparticles and influence on magnetic properties. J Phys Chem C 113(38):16681–16685
Ahrén M, Selegård L, Söderlind F, Linares M, Kauczor J, Norman P, Käll P-O, Uvdal K (2012) A simple polyol-free synthesis route to Gd 2 O 3 nanoparticles for MRI applications: an experimental and theoretical study. J Nanopart Res 14(8):1006
Di W, Ren X, Zhang L, Liu C, Lu S (2011) A facile template-free route to fabricate highly luminescent mesoporous gadolinium oxides. CrystEngComm 13(15):4831–4833
Park JY, Baek MJ, Choi ES, Woo S, Kim JH, Kim TJ, Jung JC, Chae KS, Chang Y, Lee GH (2009) Paramagnetic ultrasmall gadolinium oxide nanoparticles as advanced T 1 MRI contrast agent: account for large longitudinal relaxivity, optimal particle diameter, and in vivo T 1 MR images. ACS Nano 3(11):3663–3669
Caravan P, Ellison JJ, McMurry TJ, Lauffer RB (1999) Gadolinium (III) chelates as MRI contrast agents: structure, dynamics, and applications. Chem Rev 99(9):2293–2352
Fang J, Chandrasekharan P, Liu X-L, Yang Y, Lv Y-B, Yang C-T, Ding J (2014) Manipulating the surface coating of ultra-small Gd2O3 nanoparticles for improved T1-weighted MR imaging. Biomaterials 35(5):1636–1642
Shahbazi M-A, Hamidi M, Mäkilä EM, Zhang H, Almeida PV, Kaasalainen M, Salonen JJ, Hirvonen JT, Santos HA (2013) The mechanisms of surface chemistry effects of mesoporous silicon nanoparticles on immunotoxicity and biocompatibility. Biomaterials 34(31):7776–7789
Zhang H-W, Wang L-Q, Xiang Q-F, Zhong Q, Chen L-M, Xu C-X, Xiang X-H, Xu B, Meng F, Wan Y-Q (2014) Specific lipase-responsive polymer-coated gadolinium nanoparticles for MR imaging of early acute pancreatitis. Biomaterials 35(1):356–367
Y-k Lee (2006) Preparation and characterization of folic acid linked poly (L-glutamate) nanoparticles for cancer targeting. Macromol Res 14(3):387–393
Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP (2005) Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem 338(2):284–293
Lee D-E, Koo H, Sun I-C, Ryu JH, Kim K, Kwon IC (2012) Multifunctional nanoparticles for multimodal imaging and theragnosis. Chem Soc Rev 41(7):2656–2672
Xie J, Lee S, Chen X (2010) Nanoparticle-based theranostic agents. Adv Drug Deliv Rev 62(11):1064–1079
Mi P, Kokuryo D, Cabral H, Kumagai M, Nomoto T, Aoki I, Terada Y, Kishimura A, Nishiyama N, Kataoka K (2014) Hydrothermally synthesized PEGylated calcium phosphate nanoparticles incorporating Gd-DTPA for contrast enhanced MRI diagnosis of solid tumors. J Control Release 174:63–71
Acknowledgements
This work was supported in part by the research chancellor of Tehran University of Medical Sciences (Grant no. 96-04-30-36739), Tehran, Iran.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors do not have any conflict of interest.
Ethical approval
All in vivo protocols were performed based on the European Community guidelines and was approved by local ethical committee, Tehran University of Medical Sciences (TUMS), Tehran, Iran (Approval number: IR.TUMS.REC0.1394.1461).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mortezazadeh, T., Gholibegloo, E., Alam, N.R. et al. Gadolinium (III) oxide nanoparticles coated with folic acid-functionalized poly(β-cyclodextrin-co-pentetic acid) as a biocompatible targeted nano-contrast agent for cancer diagnostic: in vitro and in vivo studies. Magn Reson Mater Phy 32, 487–500 (2019). https://doi.org/10.1007/s10334-019-00738-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10334-019-00738-2