
Abstract
Objective
Tracking the autoreactive T-cell migration in the pancreatic region after labeling with fluorinated nanoparticles (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[3-(2-pyridyldithio)propionate]-perfluoro-15-crown-5-ether nanoparticles, PDP-PFCE NPs) in a diabetic murine model using 19F MRI.
Materials and methods
Synthesis of novel PDP-PFCE fluorine tracer was performed for in vitro labeling of T cells. Labeling conditions were optimized using different PDP-PFCE NPs concentrations. For in vivo 19F MRI, mice were longitudinally followed after adoptive transfer of activated, autoreactive, labeled T cells in NOD.SCID mice.
Results
Established MR protocols were used for challenging T cell labeling to track inflammation in a model of diabetes after successful labeling of CD4+ and CD8+ T cells with PDP-PFCE NPs. However, T cells were difficult to be detected in vivo after their engraftment in animals.
Discussion
We showed successful in vitro labeling of T cells using novel fluorinated liposomal nanoparticles. However, insufficient and slow accumulation of labeled T cells and subsequent T cell proliferation in the pancreatic region remains as limitations of in vivo cell imaging by 19F MRI.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Autoimmune type 1 diabetes (T1D) results from a decline in insulin-producing pancreatic β-cells, resulting in hyperglycemia when β-cells are selectively destroyed by the autoimmune repertoire. The diagnosis of T1D mainly includes serological fasting blood glucose measurements and identification of genetic markers. Over the past years, some progress has been made for the in vivo visualization of pancreatic β-cells in both pre-clinical and clinical settings [1,2,3,4,5]. In T1D, autoreactive T cells are the main culprits responsible for inflammation and the depletion of insulin-producing pancreatic β-cells. However, the interplay between immune cells and β-cells remains poorly understood and was only visualized by invasive imaging techniques [6, 7]. Non-invasive assessment of the onset of inflammation and autoimmunity is currently unavailable but would improve monitoring of the disease and possible treatment. Several studies have been conducted to understand the role of T cells, where both CD4+ and CD8+ T cells are found to be crucial in the induction of T1D [8, 9]. The ability to visualize immune cells after in vitro labeling with potential contrast agent could provide a powerful tool to understand the evolution of the disease and establish testing of novel therapeutic approaches. The potential value of non-invasive imaging in elucidating underlying inflammatory processes includes monitoring the dynamics of cellular interactions and disease progression [10].
In this regard, in vivo cell imaging methods have gained momentum over the last decade [11, 12]. Hereby, magnetic resonance imaging (MRI) is one of the most powerful in vivo imaging tools due to its high resolution and soft tissues contrast. For the visualization of specific cell types, MRI contrast agents are needed that are (a) highly specific to the targeted cell type, (b) are sensitive, (c) generate unambiguous contrast, (d) stably attach to/are incorporated by the cells, (e) not affecting cell biology and (f) generating longitudinal contrast changes [13].
Until now, most imaging applications for the in vivo visualization of inflammatory processes and cells of the immune system have mainly focused on models with high numbers of immune cell infiltrating at the site of injury or disease. The utilization of sensitive superparamagnetic iron oxide (SPIO) particles as contrast agents was often hampered by unfavorable background contrast, unspecific contrast origin and the inability to accurately quantify the extent of inflammation [14]. Nowadays, fluorine (19F) MRI has gained increased popularity also for the visualization of inflammatory processes where the MR signal from the 19F atoms can be directly quantified [15,16,17,18]. As a potential contrast agent for 19F MRI, perfluoro-15-crown-5-ether nanoparticles (PFCE-NPs) are promising due to the large number of equivalent fluorine atoms per molecule [19, 20]. It has been shown that, when intravenously injected, PFCE-NPs do not extravasate but are phagocytozed mainly by macrophages allowing the measurement of immune cell infiltration corresponding to the inflammation in disease models.
On the other hand, it has been extremely challenging to label non-phagocytic immune cells like T cells for their in vivo follow-up after their engraftment in a suitable T1D model [21, 22]. To overcome such challenges, chemical methods like the use of transfection agents are successfully applied to label non-phagocytic cells like T cells and visualize them in a murine model of diabetes [23]. However, it has been reported that transfection agents may induce cellular toxicity or immune activation, potentially disturbing the normal physiological function of T cells [24]. To achieve a sufficient cellular fluorine concentration for generating detectable 19F MR signal without using transfection agents, we applied a lipid modification of PFCE NPs with PDP (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[3-(2-pyridyldithio)propionate]) (PDP-PFCE NPs), facilitating the interaction of T-cell membrane with the nanoparticles [25].
In this study, we aimed to visualize islet antigen-specific (NOD-derived BDC2.5), autoreactive T cells labeled with novel PDP-PFCE NPs after adoptive transfer in NOD.SCID mice using 19F MR imaging.
Materials and methods
Animals
BDC2.5 T cell receptor (TCR)-Tg NOD mice aged 6–8 weeks (n = 8) and NOD.SCID mice aged 5–6 weeks (n = 13) were obtained from the breeding facility of Clinical and Experimental Endocrinology (KU Leuven, Leuven, Belgium). All animals were housed in micro-isolator IVC cages, fed with standard chow diet, and received tap water ad libitum. All in vivo experiments were approved by the Institutional Animal Care and ethical Committee.
Synthesis of PFCE and PDP-PFCE nanoparticles
PFCE NPs were prepared, as described previously [26]. Further, 40.2 wt% DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine), 50 wt% PE-PDP (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[3-(2-pyridyldithio)propionate]), and 9.8 wt% cholesterol (Avanti Polar Lipids, Alabaster, USA) all dissolved in chloroform was mixed with PFCE-NPs to synthesize PDP-PFCE NPs.
For flow cytometry, anionic PDP-PFCE NPs were incorporated with 2.5 wt% of the lipophilic DiR fluorescent dye (Thermo Scientific, Cramlington, UK). After evaporation of chloroform, the lipid film was hydrated in a 1:2:7 of glycerol, propanediol (Thermo Scientific) and MiliQ water to obtain total lipid concentration of 1.4 × 10−3 mmol/mL.
Isolation and activation of splenic T lymphocytes
Splenocytes obtained from BDC2.5 TCR-Tg NOD mice were maintained in RPMI 1640 medium (Invitrogen, Merelbeke, Belgium) supplemented with Glutamax (Gibco, Germany), 10% FCS, 10 mM HEPES, 1 mM glutamine, 1 mM Na-pyruvate and 50 μM 2-ME (Invitrogen, Merelbeke, Belgium). After the lysis of red blood cells using NH4Cl at room temperature, T cells were resuspended at 106 cells/mL. For T-cell activation, splenocytes were pulsed with BDC2.5 mimotope (AnaSpec, Fremont, California, USA) at a concentration of 0.1 µg/mL and incubated at 37 °C for 72 h as described [27]. Purity of T cells was assessed ~ 90–95% for individual experiments using flow cytometry.
In vitro labeling of splenic T cells with PDP-PFCE nanoparticles
After 72 h of activation, T cells were washed with serum-free media and co-incubated with DiR-coupled fluorescent PDP-PFCE nanoparticles for 3 h at 37 °C. For flow cytometry, labeled T cells were washed for the removal of free nanoparticles with FACS buffer [Phosphate Buffer Saline (PBS) with 0.1% BSA (Invitrogen, Merelbeke, Belgium) and 2 mM EDTA (Merck, Darmstadt, Germany)]. Pre-incubation with Fc receptor-blocking antibody and cell surface staining was performed using an antibody mix of anti-CD4-FITC, anti-CD8-efluor450 and CD11/b-PE. Dead cells were excluded from each analysis using Live/Dead yellow dye (Invitrogen, Merelbeke, Belgium).
Imaging of PDP-PFCE NP-labeled T cells in phantoms and in vivo using 19F MRI
Splenic T cells were labeled with PDP-PFCE NPs (total fluorine concentration: 20 mM) and embedded in centrifugation tubes (Eppendorf, Rotselaar, Belgium) containing 2% agar (Merck, Darmstadt, Germany) and 5 × 105, 1 × 106, 2.5 × 106 or 5 × 106 cells (n = 3), respectively. To mimic the in vivo environment, the same amount of labeled T cells suspended in 100 µl sterile PBS were subcutaneously engrafted in mice before acquiring 19F MR images. 19F MRI was performed within 30–60 min following cell engraftment.
19F MR imaging of labeled T cells
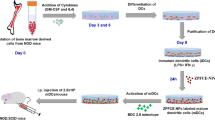
All 19F/1H MR images were acquired on a preclinical 9.4T Bruker Biospec MR scanner (Bruker Biospec 94/20, Ettlingen, Germany). MR imaging was performed with a purpose-built dual-tuned surface coil for 1H (proton) and 19F (fluorine) MR using the same orientation and placement of slice packages. For the monitoring of T cells, a first recipient group of NOD.SCID mice (n = 5) received 1 × 106 and a second recipient group of NOD.SCID mice (n = 5) received 5 × 106 BDC2.5-mimotope-loaded PDP-PFCE-labeled T cells via intravenous injections, (Fig. 1).

Experimental timeline for the in vitro labeling and in vivo 19F MR imaging after adoptive transfer of T cells in a T1D mouse model
The control group received saline. For the T1D model, MR images were acquired in coronal orientation. For all acquisitions, a RARE (Rapid Acquisition with Relaxation Enhancement) MR sequence was used with the following parameters: anatomical images were acquired with TE = 6.121 ms, RARE factor = 8, TR = 3500 ms, spatial resolution = 0.156 mm × 0.156 mm, slice thickness 1 mm. Fluorine images were acquired with TE = 6.11 ms, RARE factor = 32, TR = 5388 ms, spatial resolution = 1.25 mm × 1.25 mm, slice thickness = 2 mm.
For phantom experiments, anatomical images with TE = 6.121 ms, RARE factor = 8, TR = 3500 ms, spatial resolution = 0.175 mm × 0.175 mm, slice thickness 1 mm were acquired. Parameters used for fluorine images were: TE = 6.11, rare factor = 32, TR = 4500 ms, spatial resolution = 1.406 mm × 1.406 mm, slice thickness = 2 mm.
In vivo estimation of fluorine concentrations in various organs
For the data processing, 1H MR images were overlaid on the 19F MR images using MeVislab software version 2.6.1 (MeVis Medical Solutions AG, Bremen, Germany). A reference tube with known number of 19F atoms was placed next to the liver of all animals. With the help of Gaussian smoothing and interval thresholding, 19F MR images were masked as described previously [26]. The regions of interest from 19F MR images were manually delineated to obtain mean signal intensities relative to the reference for the quantification of fluorine atoms.
Ex vivo validation
For the T1D model, pancreases were isolated and prepared for flow cytometry from all NOD.SCID mice groups on day 9, after in vivo transfer of activated labeled T cell, as described previously [27]. Briefly, pancreatic tissue was finely minced and suspended in RPMI complete medium containing 1 mg/mL collagenase VIII and 0.02 mg/mL DNAase I (Merck, Darmstadt, Germany) for tissue digestion. Single cell suspensions were obtained from homogenized tissues and strained using a 70 µm nylon strainer (both from Miltenyi Biotec, Leiden, The Netherlands). Cells were surface-stained for flow cytometry using CD4 + and CD8 + T cell antibodies, for the identification of PDP-PFCE NP-labeled T cells.
Flow cytometry and confocal microscopy
All antibodies used for flow cytometry were purchased from eBioscience (San Diego, California, USA). Flow cytometry was performed on a Gallios flow cytometer (Beckman Coulter, Analis, Suarlèe, Belgium), and data analyses were executed on FlowJo software®, (version 10.4.2, FlowJo LLC, Ashland, Oregon, USA). Confocal microscopy was performed on a Nikon Eclipse A1R microscope (Nikon, Tokyo, Japan).
19F nuclear magnetic resonance (NMR) spectroscopy of cell suspensions
To assess the amount of fluorine label per cell, either 2.5 × 105, 5 × 105, 1 × 106, 2.5 × 106 or 5 × 106 PDP-PFCE-labeled T cells were embedded in agar-filled Eppendorf tubes and imaged as described above or 19F NMR experiments were performed using a 400 MHz NMR spectrometer with an Avance II console (Bruker Biospin GmbH, Rheinstetten, Germany) as described before [1]. For the latter, cells were suspended in 5 mm NMR tubes (Wilmad, Vineland, NJ, USA). A 5 mm broadband probe was used with an operating frequency for 19F NMR of 376.50 MHz. For all 19F NMR experiments, the following parameters were used: relaxation delay: 5 s, number of acquisitions: 1024, spectral width: 350 ppm and 128 k data points. NMR data analyses/quantifications were performed using the TopSpin software (Bruker Biospin, Rheinstetten, Germany) by integrating 19F NMR signals after phase and baseline corrections and comparison to an internal standard of known concentration. As a reference compound, 5-fluorocytosine or sodium fluoride (0.1 mL, 5 mM 19F concentration) was added to determine the chemical shift and 19F concentration.
Statistical analysis
Data sets were analyzed using one-way ANOVA and Bonferroni’s multiple comparison tests with Prism (version 6, GraphPad software, San Diego, USA). Data are represented as mean ± SEM.
Results
In vitro labeling of NOD/BDC2.5 transgenic T cells with PDP-PFCE nanoparticles
Islet antigen-specific autoreactive T cells isolated from the spleen of NOD/BDC2.5 TCR Tg mice were isolated as described [28]. T cells were activated with BDC 2.5 mimotope and labeled with PDP-PFCE NPs. Flow cytometry was performed for quantitative analysis of labeled T cells at 10 and 20 mM PDP-PFCE NPs concentrations (Fig. 2).

Quantitative and qualitative analyses of T cells labeled with PDP-PFCE nanoparticles. a Specific T cell markers were used for the identification of CD4+ and CD8+ T cell populations. b Histogram representation of PDP-PFCE NP-labeled T cells at concentrations of 10 and 20 mM, respectively. c Percentage of labeled CD4+ and CD8+ T cells and the median fluorescence intensities (MFI) were measured for fluorine concentrations up to 20 mM PDP-PFCE using flow cytometery (n = 3, mean ± SEM, ****P < 0.0001). d Qualitative confocal microscopy, showing CD4+ T cells labeled with PDP-PFCE NPs at the concentration of 20 mM, (blue = nuclei, red = PDP-PFCE NPs, green = CD4+ T cell marker, scale bar = 20 µm)
Both CD4+ and CD8+ viable T cells were analyzed for fluorine concentrations of 10 and 20 mM to assess the association with PDP-PFCE NPs (Fig. 2a). A threshold median fluorescence intensity (MFI) was applied by implementing T cells without PDP-PFCE NPs (Fig. 2b). Consequently, the frequency of PDP-PFCE labeled cells and particles/cell was determined following exposure to two different particle concentrations (Fig. 2c). Maximal T cell labeling with PDP-PFCE NPs was observed at a fluorine concentration of 20 mM. For the qualitative analysis of the PDP-PFCE NP-labeled T cells and further confirmation of label association, T cells were analyzed by confocal microscopy using a fluorine concentration of 20 mM (Fig. 2d).
19F MR imaging and NMR spectroscopy of PDP-PFCE NP-labeled T cells
To estimate the detectable number of PDP-PFCE NP-labeled T cells, phantoms that contained different numbers of labeled T cells were prepared after labeling T cells with PDP-PFCE NPs using a total fluorine concentration of 20 mM. As expected, a linear correlation was observed between the 19F MRI signal intensity with the number of PDP-PFCE NP-labeled T cells (Fig. 3a, b). The lowest detectable amount of T cells was approximately 5 × 105. The signal-to-noise ratio (SNR) for 5 × 105 PDP-PFCE NP labeled T cells in phantoms was 18. Results obtained from 19F NMR spectroscopy confirmed linear correlation between the number of 19F spins with the amount of labeled T cells (Fig. 3c). Based on 19F NMR spectroscopy, T cells contained on average 7 ± 2 × 1012 fluorine atoms/cell. To test the detection limits of PDP-PFCE-labeled T cells in vivo, we subcutaneously engrafted different amounts of labeled T cells subcutaneously in healthy mice. Based on the in vivo 19F MR imaging, we were also able to detect down to 5 × 105 labeled T cells with an SNR of approximately 8 (Fig. 3d, e).

In vitro and in vivo 19F MR cell imaging and estimation of fluorine spins of PDP-PFCE NP-labeled T cells. a, b Overlay of fluorine signal obtained from phantom with different amounts of PDP-PFCE NP-labeled T cells and representation of the correlation between fluorine concentration and the number of PDP-PFCE NP-labeled T cells at different cell concentrations (R = 0.9881), respectively. c19F NMR spectroscopy-based quantification of fluorine atoms from T cell (R = 0.9856). The signal intensity of PDP-PFCE NPs was compared to an internal chemical shift and concentration reference. d, e Subcutaneous implantation of different concentrations of PDP-PFCE-labeled T cells in mice, for the quantification of fluorine signal and estimation of detection threshold (R = 0.9229). The PDP-PFCE concentration used for labeling T cells was 20 mM. 1 = 5 × 105, 2 = 1 × 106, 3 = 2.5 × 106 and 4 = 5 × 106
In vivo assessment of autoreactive T cells labeled with PDP-PFCE NPs post-adoptive transfer in NOD.SCID mice
Autoreactive T cells were loaded with BDC2.5 mimotope and labeled with 20 mM PDP-PFCE nanoparticles. The adoptive transfer of PDP-PFCE NP-labeled T cells in NOD.SCID mice was performed via intravenous injection where group 1 and 2 received 1 × 106 and 5 × 106 T cells, respectively. The control group did not receive T cells.
After 24 h following adoptive transfer of PDP-PFCE NPs labeled T cells, 19F MR images were acquired from all groups of NOD.SCID mice for up to 8 days. We did not observe any 19F MRI-detectable fluorine signal from the pancreatic or other regions of all groups of NOD.SCID mice at any of the 19F MR imaging time-points (Fig. 4).

Longitudinal in vivo MR imaging to follow-up PDP-PFCE NP-labeled T cells after adoptive transfer in a T1D model. All groups of animals were monitored by acquiring 1H and 19F MR images of the pancreatic regions from day 1 to day 8. The images shown above are from a representative animal that has received 5 × 106 labeled T cells. A fluorine reference (30 mM) was placed next to the animal for the quantification of 19F signal
Determination of T1D onset and ex vivo quantification of T cells in the pancreas
To verify whether transferred labelled T cells were functional in vivo and the loss of label was not because of cell death or dysfunctional T cells, we monitored blood glucose level and performed ex vivo flow cytometry. The onset of T1D was determined by recording the blood glucose levels on all non-fasted NOD.SCID animals from day 1 until day 12, post adoptive transfer of PDP-PFCE-labeled T cells. As expected, animals receiving 5 × 106 labeled T cells showed an earlier onset of T1D (day 6) than animals receiving 1 × 106 T cells (day 10), as shown in Fig. 5a. The control group featured normal blood glucose concentrations.

Onset of type-1 diabetes and ex vivo flow cytometry of the pancreas. a After the labeled T cells engraftment, blood glucose level was measured for up to 12 days from non-fasted NOD.SCID animals to determine the onset of T1D. b Ex vivo flow cytometry on pancreases, showing the presence of CD4+ T cells detected on day 12 post transplantation in both mice groups (mean ± SEM, *P < 0.05)
To quantify the number of migrating PDP-PFCE NP-labeled T cells in the pancreas, we performed ex vivo flow cytometry on day 12. We did not observe PDP-PFCE NP-labeled T cells as confirmed by flow cytometry. However, CD4+ T cells were observed in both the recipient NOD.SCID groups with a higher percentage in the group receiving more cells (Fig. 5b). This shows that the T cells were functional and effectively migrated to the pancreas although, no association of PDP-PFCE NPs was observed with the isolated T cells.
In vitro assessment of PDP-PFCE label dilution in T cells
To explain the low degree of T cell labeling in the pancreatic region, we have assessed the dilution of PDP-PFCE NP-labeling of T cells with continued proliferation in vitro. Hereby, 5 × 106 T cells were labeled with 20 mM PDP-PFCE nanoparticles and labeling was assessed at different time-points using flow cytometry. We found a decrease in the number of CD4+ and CD8+ T cells labeled with PDP-PFCE nanoparticles after 24 h (Fig. 6a, c). The median fluorescence intensities showed that the number of PDP-PFCE nanoparticles associated with T cells decreased rapidly over time (Fig. 6b–d).

In vitro assessment of PDP-PFCE labeling on BDC 2.5-activated T cells. ad–d Quantification of the labeling and MFI of CD4+ and CD8+ T cells with PDP-PFCE particles (20 mM fluorine concentration) for assessment of the label dilution for up to 48 h, respectively (mean ± SEM)
Discussion
In T1D, the breach of tolerance mechanisms results in the emergence of self-reactive T cells, which infiltrate the islets along with monocytes/macrophages and destroy the pancreatic β-cells [9, 29]. As inflammation is a hallmark of autoimmune diseases like T1D, it is important to assess the onset and progression of these immunological processes. The high contrast specificity of 19F MR imaging when compared to the use of SPIO agents is hence a promising imaging tool, which has been exploited for in vitro and in vivo visualization of tracer-labeled immune cells and to quantify inflammation [16, 23, 30,31,32]. In this study, we employed adoptive transfer of novel PDP-PFCE tracer-labeled autoreactive T cells into immunodeficient T1D-susceptible animals. We followed the T cell recipient NOD.SCID mice aiming to visualize pancreatic inflammation by longitudinal 19F MRI.
Delivery of magnetic nanoparticles (MNPs) in vivo has already been successful in demonstrating changes in the pancreatic vasculature of diabetic mouse models, after accumulation of labeled nanoparticles in infiltrating macrophages [33]. However, labeling of non-phagocytic immune cells like T cells is challenging, as they do not follow the endocytic pathway for cellular uptake. While transfection agents are readily used to enhance the labeling of T cells also for 19F MRI-based tracking [23], chemical methods may affect physiological immune function of labeled T cells [34]. Other large scale industrial processes were also exploited to label T cells using commercially available 19F tracers for monitoring T cell immunotherapy; however, this procedure is difficult to implement for small-scale laboratory experiments [35]. We therefore assessed whether lipid modification of PFCE-containing nanoparticles can facilitate their interaction with the T cell membrane, where we successfully labeled CD4+ and CD8+ T cells in vitro with PDP-PFCE nanoparticles. Comparison with previous attempts to label T cells with fluorine-containing contrast agents for in vivo monitoring showed labeling efficiencies between 1 and 22 × 1012 fluorine atoms per cell [22, 23]. Without using transfection agents, we were able to achieve T cell labeling in a similar concentration range (7 × 1012 fluorine atoms per T cell). From our 19F MR imaging data of phantoms, we were able to quantify T cells in a range from 0.5 to 5 million T cells labeled with PDP-PFCE NPs, potentially suitable for accurate and quantitative cell tracking.
However, in vivo 19F MR images showed no detectable fluorine signal from the pancreatic region of NOD.SCID recipient mice, engrafted with different amounts of labeled T cells. Transplanted animals became diabetic 5 days after adoptive transfer of labeled T cells, indicating that the engrafted T cells were able to migrate to the pancreas for the destruction of β-cells. Further, based on our in vitro PDP-PFCE label dilution experiments, we can conclude that the PDP-PFCE label was probably diluted due to T cell proliferation in the pancreatic region. Based on these findings, T cells were sufficiently labeled with PDP-PFCE NPs for up to 48 h, but the number of particles/cell was not sufficient to achieve detectable in vivo 19F MR signal after sufficient local accumulation of T cells in the pancreatic region 5 days after engraftment. As measured by the 19F MRI experiments of phantoms, the detectability threshold was estimated to be ~ 1016 fluorine atoms/pixel, which is comparable with previous publications [36, 37]. This is sufficient for models of relative rapid local in vivo accumulation of labeled cells like the LPS model [15], but the in vivo detection of PDP-PFCE NP-labeled T cells in a T1D model remains elusive and challenging using 19F MR imaging. This illustrates the ‘biological’ requirements for making in vivo 19F MRI-based cell imaging successful: (1) sufficiently high amounts of locally accumulated labeled cells are needed, and (2) cell proliferation and associated label dilution should be minimal.
If these ‘biological’ requirements cannot be met, ‘technical’ improvements are needed like the development of more efficient fluorinated nanoparticles to achieve labeling of T cells, that is sufficient for in vivo monitoring by 19F MRI. Synthesis of fluorine tracers with high number of fluorine atoms is required to compensate for the dilution after the cell divisions. To avoid the utilization of tranfection agents, which have been used with fluorinated contrast agents for in vivo monitoring of T cells [23] we have used NPs with a modified surface (PDP-PFCE NPs). Although, we have achieved almost as high cellular fluorine concentrations as have been achieved with transfection agents [23] we failed to monitor T cell infiltration in vivo. Considering the efforts also taken by other research groups, this is not completely unexpected [22]. Using terminally divided T cells or higher numbers of engrafted T cells than routinely used in this model are ‘biological’ modifications for achieving high in vitro labeling and in vivo visualization upon transfer in T1D model. However, as proliferation of T cells is their characteristic feature for the generation of adaptive immune response, such a model would be irrelevant to study inflammation in this disease model.
In conclusion, the utilization of transfection agents is currently the only approach that has resulted in sufficiently high fluorine concentrations for in vivo monitoring of T cells. As the utilization of transfection agents for diagnostic imaging in the clinic is currently not approved, other ways for achieving sufficient sensitivity of in vivo 19F MRI of non-phagocytic cells may include more sensitive and dedicated 19F coils like cryogen-cooled coils and improved processing methods like compressed sensing techniques [38, 39]. However, it remains to be seen if these improvements will be sufficient to overcome the detectability limitations of 19F MRI cell imaging applications for non-phagocytic cells. These improvements will further aid in the translation of preclinical methods to the clinical settings.
References
Liang S, Louchami K, Kolster H, Jacobsen A, Zhang Y, Thimm J, Sener A, Thiem J, Malaisse W, Dresselaers T, Himmelreich U (2016) In vivo and ex vivo 19-fluorine magnetic resonance imaging and spectroscopy of beta-cells and pancreatic islets using GLUT-2 specific contrast agents. Contrast Media Mol Imaging 11:506–513. https://doi.org/10.1002/cmmi.1712
Kriz J, Jirak D, Berkova Z, Herynek V, Lodererova A, Girman P, Habart D, Hajek M, Saudek F (2012) Detection of pancreatic islet allograft impairment in advance of functional failure using magnetic resonance imaging. Transpl Int 25:250–260. https://doi.org/10.1111/j.1432-2277.2011.01403.x
Malosio ML, Esposito A, Brigatti C, Palmisano A, Piemonti L, Nano R, Maffi P, De Cobelli F, Del Maschio A, Secchi A (2015) MR imaging monitoring of iron labeled pancreatic islets in a small series of patients: islets fate in successful, unsuccessful and auto-transplantation. Cell Transplant 24:2285–2296. https://doi.org/10.3727/096368914X684060
Medarova Z, Moore A (2008) Non-invasive detection of transplanted pancreatic islets. Diabetes Obes Metab 10(Suppl 4):88–97. https://doi.org/10.1111/j.1463-1326.2008.00942.x
Arifin DR, Bulte JWM (2011) Imaging of pancreatic islet cells. Diabetes Metab Res Rev 27:761–766. https://doi.org/10.1002/dmrr.1248
Alanentalo T, Asayesh A, Morrison H, Lorén CE, Holmberg D, Sharpe J, Ahlgren U (2007) Tomographic molecular imaging and 3D quantification within adult mouse organs. Nat Methods 4:31–33. https://doi.org/10.1038/nmeth985
Alanentalo T, Lorén CE, Larefalk A, Sharpe J, Holmberg D, Ahlgren U (2008) High-resolution three-dimensional imaging of islet-infiltrate interactions based on optical projection tomography assessments of the intact adult mouse pancreas. J Biomed Opt 13:054070. https://doi.org/10.1117/1.3000430
Peterson JD, Haskins K (1996) Transfer of diabetes in the NOD-scid mouse by CD4 T-cell clones: differential requirement for CD8 T-cells. Diabetes 45:328–336
Wucherpfennig KW, Eisenbarth GS (2001) Type 1 diabetes. Nature 2:767–768. https://doi.org/10.1016/S0140-6736(11)60614-4
Srinivas M, Boehm-Sturm P, Figdor CG, de Vries IJ, Hoehn M (2012) Labeling cells for in vivo tracking using 19F MRI. Biomaterials 33:8830–8840. https://doi.org/10.1016/j.biomaterials.2012.08.048
Himmelreich U, Hoehn M (2008) Stem cell labeling for magnetic resonance imaging. Minim Invasive Ther Allied Technol 17:132–142. https://doi.org/10.1080/13645700801969873
Himmelreich U, Dresselaers T (2009) Cell labeling and tracking for experimental models using Magnetic Resonance Imaging. Methods 48:112–124. https://doi.org/10.1016/j.ymeth.2009.03.020
Srinivas M, Heerschap A, Ahrens ET, Figdor CG, de Vries IJM (2010) 19F MRI for quantitative in vivo cell tracking. Trends Biotechnol 28:363–370. https://doi.org/10.1016/j.tibtech.2010.04.002
Harms C, Datwyler AL, Wiekhorst F, Trahms L, Lindquist R, Schellenberger E, Mueller S, Schütz G, Roohi F, Ide A, Füchtemeier M, Gertz K, Kronenberg G, Harms U, Endres M, Dirnagl U, Farr TD (2013) Certain types of iron oxide nanoparticles are not suited to passively target inflammatory cells that infiltrate the brain in response to stroke. J Cereb Blood Flow Metab 36(Suppl 1):139–140. https://doi.org/10.1038/jcbfm.2013.22
Ebner B, Behm P, Jacoby C, Burghoff S, French BA, Schrader J, Flögel U (2010) Early assessment of pulmonary inflammation by 19F MRI in vivo. Circ Cardiovasc Imaging 3:202–210. https://doi.org/10.1161/CIRCIMAGING.109.902312
Stoll G, Basse-Lüsebrink T, Weise G, Jakob P (2012) Visualization of inflammation using 19F-magnetic resonance imaging and perfluorocarbons. Wiley Interdiscip Rev Nanomedicine Nanobiotechnology 4:438–447. https://doi.org/10.1002/wnan.1168
Shin SH, Kadayakkara DK, Bulte JWM (2017) In Vivo 19 F MR imaging cell tracking of inflammatory macrophages and site-specific development of colitis-associated dysplasia. Radiology 282:194–201. https://doi.org/10.1148/radiol.2016152387
Jacoby C, Borg N, Heusch P, Sauter M, Bönner F, Kandolf R, Klingel K, Schrader J, Flögel U (2014) Visualization of immune cell infiltration in experimental viral myocarditis by 19F MRI in vivo. Magn Reson Mater Phy. https://doi.org/10.1007/s10334-013-0391-6
Tirotta I, Dichiarante V, Pigliacelli C, Cavallo G, Terraneo G, Bombelli FB, Metrangolo P, Resnati G (2015) 19F magnetic resonance imaging (MRI): from design of materials to clinical applications. Chem Rev 115:1106–1129
Janjic JM, Ahrens ET (2009) Fluorine-containing nanoemulsions for MRI cell tracking. Wiley Interdiscip Rev Nanomedicine Nanobiotechnology 1:492–501
Westermann J, Söllner S, Ehlers E-M, Nohroudi K, Blessenohl M, Kalies K (2003) Analyzing the migration of labeled T cells in vivo: an essential approach with challenging features. Lab Investig 83:459–469. https://doi.org/10.1097/01.LAB.0000062852.80567.90
Gonzales C, Yoshihara HAI, Dilek N, Leignadier J, Irving M, Mieville P, Helm L, Michielin O, Schwitter J (2016) In-vivo detection and tracking of T cells in various organs in a melanomatumor model by 19F-fluorine MRS/MRI. PLoS One 11:1–18. https://doi.org/10.1371/journal.pone.0164557
Srinivas M, Morel PA, Ernst LA, Laidlaw DH, Ahrens ET (2007) Fluorine-19 MRI for visualization and quantification of cell migration in a diabetes model. Magn Reson Med 58:725–734. https://doi.org/10.1002/mrm.21352
Przybylski S, Gasch M, Marschner A, Ebert M, Ewe A, Helmig G, Hilger N, Fricke S, Rudzok S, Aigner A, Burkhardt J (2017) Influence of nanoparticle-mediated transfection on proliferation of primary immune cells in vitro and in vivo. PLoS One 12:1–16. https://doi.org/10.1371/journal.pone.0176517
Wayteck L, Dewitte H, De Backer L, Breckpot K, Demeester J, De Smedt SC, Raemdonck K (2016) Hitchhiking nanoparticles: reversible coupling of lipid-based nanoparticles to cytotoxic T lymphocytes. Biomaterials 77:243–254. https://doi.org/10.1016/j.biomaterials.2015.11.016
Liang S, Louchami K, Holvoet B, Verbeke R, Deroose CM, Manshian B, Soenen SJ, Lentacker I, Himmelreich U (2018) Tri-modal in vivo imaging of pancreatic islets transplanted subcutaneously in mice. Mol Imaging Biol 20:940–951. https://doi.org/10.1007/s11307-018-1192-0
Ferreira GB, Gysemans CA, Demengeot J, da Cunha JPMCM, Vanherwegen A-S, Overbergh L, Van Belle TL, Pauwels F, Verstuyf A, Korf H, Mathieu C (2014) 1,25-Dihydroxyvitamin D3 promotes tolerogenic dendritic cells with functional migratory properties in NOD mice. J Immunol 192:4210–4220. https://doi.org/10.4049/jimmunol.1302350
Poulin M, Haskins K (2000) Induction of diabetes in nonobese diabetic mice by Th2 T cell clones from a TCR transgenic mouse. J Immunol 164:3072–3078
Eizirik DL, Colli ML, Ortis F (2009) The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol 5:219–226. https://doi.org/10.1038/nrendo.2009.21
Ahrens ET, Bulte JWM (2013) Tracking immune cells in vivo using magnetic resonance imaging. Nat Rev Immunol 13:755–763
van Heeswijk RB, Pellegrin M, Flögel U, Gonzales C, Aubert J-F, Mazzolai L, Schwitter J, Stuber M (2015) Fluorine MR imaging of inflammation in atherosclerotic plaque in vivo. Radiology 275:421–429. https://doi.org/10.1148/radiol.14141371
Temme S, Bönner F, Schrader J, Flögel U (2012) 19F magnetic resonance imaging of endogenous macrophages in inflammation. Wiley Interdiscip Rev Nanomedicine Nanobiotechnology 4:329–343. https://doi.org/10.1002/wnan.1163
Gaglia JL, Guimaraes AR, Harisinghani M, Turvey SE, Jackson R, Benoist C, Mathis D, Weissleder R (2011) Noninvasive imaging of pancreatic islet inflammation in type 1A diabetes patients. J Clin Invest 121:442–445. https://doi.org/10.1172/JCI44339
Chapelin F, Gao S, Okada H, Weber TG, Messer K, Ahrens ET (2017) Fluorine-19 nuclear magnetic resonance of chimeric antigen receptor T cell biodistribution in murine cancer model. Sci Rep 7:1–5. https://doi.org/10.1038/s41598-017-17669-4
O’Hanlon CF, Fedczyna T, Eaker S, Shingleton WD, Helfer BM (2017) Integrating a 19F MRI tracer agent into the clinical scale manufacturing of a T-cell immunotherapy. Contrast Media Mol Imaging 201:1–7. https://doi.org/10.1155/2017/9548478
Amiri H, Srinivas M, Veltien A, van Uden MJ, de Vries IJM, Heerschap A (2015) Cell tracking using 19F magnetic resonance imaging: technical aspects and challenges towards clinical applications. Eur Radiol 25:726–735. https://doi.org/10.1007/s00330-014-3474-5
Boehm-Sturm P, Mengler L, Wecker S, Hoehn M, Kallur T (2011) In Vivo tracking of human neural stem cells with 19F magnetic resonance imaging. PLoS One 6:e29040. https://doi.org/10.1371/journal.pone.0029040
Waiczies S, Millward JM, Starke L, Delgado PR, Huelnhagen T, Prinz C, Marek D, Di Wecker, Wissmann R, Koch SP, Boehm-Sturm P, Waiczies H, Niendorf T, Pohlmann A (2017) Enhanced fluorine-19 MRI sensitivity using a cryogenic radiofrequency probe: technical developments and ex vivo demonstration in a mouse model of neuroinflammation. Sci Rep 7:1–10. https://doi.org/10.1038/s41598-017-09622-2
Liang S, Dresselaers T, Louchami K, Zhu C, Liu Y, Himmelreich U (2017) Comparison of different compressed sensing algorithms for low SNR 19F MRI applications—imaging of transplanted pancreatic islets and cells labeled with perfluorocarbons. NMR Biomed 30:e3776. https://doi.org/10.1002/nbm.3776
Acknowledgements
The authors are grateful for financial support by the European Commission for the FP7 MC-ITN ‘BetaTrain’ (EU-FP7/207-2013/ 289932), by the European ERA-NET project ‘CryptoView’ (3rd call of the FP7 programme Infect-ERA), by the Flemish Wetenschap Onderzoek (FWO) for the projects G.0B28.14 and G.0A75.14, by the Agentschap Innoveren & Ondernemen for the IWT-SBO ‘NanoComit’ (140061).
Author information
Authors and Affiliations
Contributions
Study conception and design: SS, HK, IL, UH. Methodology: SL, HK, RV, KR, CG. Experimentation: SS, HK, BM, RV, SL. Analysis and interpretation of data: SS, HK, BM, UH. Drafting of manuscript: SS. Critical revision: HK, CG, IL, SD, UH.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors. Experiments involving mice were performed in accordance with regional, national and international standards on animal welfare, in particular the European Union Directive 2010/63/EU, and approved and overseen by the Animal Care and Ethical Committees of the University of Leuven.
Rights and permissions
About this article

Cite this article
Saini, S., Korf, H., Liang, S. et al. Challenges for labeling and longitudinal tracking of adoptively transferred autoreactive T lymphocytes in an experimental type-1 diabetes model. Magn Reson Mater Phy 32, 295–305 (2019). https://doi.org/10.1007/s10334-018-0720-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10334-018-0720-x