
Abstract
Sweet potato is one of the most important food crops worldwide, and viral diseases can cause very severe yield loss and quality decline of this crop. To track the prevalence of viral diseases of sweet potato in the field, 219 samples were collected from 10 locations in southern China and analysed by small RNA deep sequencing and PCR/RT-PCR. The results showed that 78.54% of the samples were infected with viruses, and 11 virus species were identified. Sweet potato virus G and sweet potato feathery mottle virus had the highest incidence and were detected in more than 40% of the samples that were tested. Coinfection of sweet potato viruses was common in southern China, and 48% of the samples were simultaneously infected by two or more virus species. These results indicate that sweet potato viruses are prevalent in southern China and highlight the importance of accurate diagnostics for their detection and the requirement for the production of virus-free plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sweet potato [Ipomoea batatas (L.) Lam.] is one of the most important crops for human consumption and animal feed worldwide and grown in all tropical and subtropical areas of the world (Clark et al. 2012). The global planting area of sweet potato is about 9 million hectares, yielding about 130 million metric tons per year. The most intensive areas of production are in southern China and around the Great Lakes of East Africa (Clark et al. 2012). Vegetative propagation, in the absence of pathogen-free material and vector control, promotes the transmission of pathogens, particularly viruses in sweet potato planting stock, resulting in yield loss and quality decline (Gibson et al. 1997; Hahn 1979; Loebenstein 2012).
Sweet potato viruses are widely distributed all over the world, including all major production areas of sweet potato. So far, over 30 virus species, assigned to nine families, that infect sweet potato have been identified (Albuquerque et al. 2012; Clark et al. 2012; Moyer and Salazar 1989). At least 15 sweet potato viruses have been reported in China, including Sweet potato chlorotic fleck virus (SPCFV), Sweet potato feathery mottle virus (SPFMV), Sweet potato virus C (SPVC), Sweet potato virus G (SPVG), Sweet potato virus 2 (SPV2), Sweet potato chlorotic stunt virus (SPCSV), Sweet potato mild mottle virus (SPMMV), Sweet potato mild speckling virus (SPMSV), Sweet potato latent virus (SPLV), Sweet potato vein mosaic virus (SPVMV), Cucumber mosaic virus (CMV), Tobacco mosaic virus (TMV), Tobacco streak virus (TSV), Sweet potato leaf curl virus (SPLCV), and Sweet potato golden vein virus (SPGVV) (Gu et al. 2014; Zhang et al. 2009). Most of these viruses are transmitted by aphids and/or whiteflies (Loebenstein 2012; Luan et al. 2007; Wang et al. 2010), and the high temperature and drought in southern China are conducive to the population growth of these two insects, thus promoting the spread of sweet potato viruses (Zhang et al. 2009).
The lack of systematic identification and detailed description of occurrence and distribution of sweet potato viruses in southern China has been a critical issue for production. In this study, 219 sweet potato samples were collected from 10 areas in southern China. Deep sequencing of virus-derived small interfering RNA (vsiRNA) was used to identify the viruses, and their presence was confirmed by PCR or RT-PCR. The results provide a basis for developing strategies to prevent the spread of sweet potato viruses and cultivating virus-free sweet potato seedlings.
Materials and methods
Collection of sweet potato samples
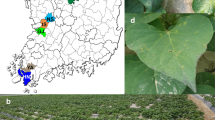
A total of 219 sweet potato samples were randomly collected from 10 locations in three provinces (Guangdong, Guangxi and Hainan) of southern China in October 2015 and October 2016 (Fig. 1). Some of the sweet potato samples had obvious symptoms such as chlorotic mottle, bright vein, purple mottle, shrinkage, deformity and mosaic leaves (Fig. 2). All samples were planted in an insect-proof greenhouse, and the leaves were collected for further analysis.

Map of 10 sampling locations in Guangdong, Guangxi and Hainan provinces in southern China. Each lettered point represents a city where the samples were collected: a, Meizhou; b, Guangzhou; c, Luoding; d, Zhanjiang; e, Hezhou; f, Liuzhou; g, Nanning; h, Tunchang; i, Lingshui; j, Baoting

Typical symptoms on field samples. a Purple spots, b shrinkage and yellowing, c chlorotic spots and yellow veins, d dwarfed, deformed leaves with mosaic, e deformed leaves with mosaic, f purple mottle, g dwarfed, deformed leaves, h pinnate mottling, i healthy leaves
Small interfering RNA sequencing and analysis
The leaf samples were pooled for vsiRNA sequencing and analysis. Total RNAs were extracted using Trizol Reagent following the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). Then total small RNAs ranging from 18 to 28 nucleotides (nt) were excised and ligated to adaptors. A small RNA library was constructed and sequenced on a HiSeq 2500 instruments (Illumina, San Diego, CA, USA). After the adaptor sequences were trimmed, 18–28-nt short reads were collected and analyzed as described by Wu et al. (2010). Briefly, de novo assembly of the small RNA reads was done using the Velvet program with 17 nt as the minimal overlapping length (k-mer) required for joining two siRNAs into a contiguous sequence (contig) (Zerbino and Birney 2008). These assembled contigs were then aligned with the BLASTN program using standard parameters for genome assembly (contigs with ≥ 90% similarity) to identify viruses.
PCR and RT-PCR
Total DNA and RNA, respectively, was extracted from each of the 219 sweet potato samples using a DNA or RNA extraction kit (Tiangen, Beijing, China). Each of the 219 samples was then tested by PCR and RT-PCR for 15 viruses, including the viruses found by deep sequencing, that have been reported as infecting sweet potato in southern China (Table S1). All primers used are listed in Table S1, and to ensure the specificity of PCR amplification, we set the annealing temperature at 55 °C. For detection of DNA viruses, PCR was performed using 2 × Taq Master Mix and the manufacturer’s instructions (Vazyme, Nanjing, Jiangsu, China). For detection of RNA viruses, one step RT-PCR was performed using a HiScript II kit and the manufacturer’s instructions (Vazyme).
Results
Main viruses infecting sweet potato in southern China
Deep sequencing of sRNAs generated 11,395,753,960 usable reads, which were assembled into 6679 contigs; 654 contigs mapped to and had 88% or more homology to virus sequences. Seven sweet potato viruses were detected in the mixed sample, including five RNA viruses, SPCFV, SPFMV, SPVC, SPVG, SPLV, and two DNA viruses, SPLCV, SPGVV. The proportion of the genome recovered for SPCFV, SPFMV, SPVC, SPVG, SPLV, SPLCV and SPGVV ranged from 9 to 70% (Table 1). This information on the main viruses in sweet potato in southern China will support further investigation of the distribution of these viruses.
Confirmation of deep sequencing data
To confirm the small RNA deep sequencing data, we used RT-PCR and PCR to detect the RNA virus and DNA viruses, respectively, from the mixed sample. Specific primers for each virus were designed based on the contig sequences from the deep sequencing data (Table S1). All seven RNA viruses (Fig. 3a) and the two DNA viruses (Fig. 3b) were amplified by RT-PCR and PCR, respectively. Sequencing of the specific bands showed they were highly homologous to the virus genomic sequences. These results further confirmed the viruses infecting sweet potato in Southern China and demonstrated the reliability of the vsiRNA deep sequencing data.

Confirmation of sweet potato viruses in the mixed sample. a, One-step RT-PCR detection of seven RNA viruses from total RNA. b, PCR detection of two DNA viruses from total DNA
Distribution of sweet potato viruses in southern China
To better describe the distribution of sweet potato virus in the region, we tested each sample for the 15 viruses reported in sweet potato, including the seven viruses found in the deep sequencing, using PCR or RT-PCR for (Table S1). Viruses were not detected in 21.46% of the 219 samples (Table 2), and 11 virus species were detected among the remaining samples, with potyviruses the most frequently detected. Potyvirus species SPVG and SPFMV had the highest incidence, in 62.10% and 40.18%, respectively, of the infected samples (Table 2). The two viruses with a very broad host range, CMV and TMV, had the lowest incidence. SPV2, SPCSV, SPVMV and TSV were not detected. The samples collected in Luoding had the most uninfected samples (84%), and only SPCFV was detected in this region. More than 10 virus species were detected in samples from Meizhou, Zhanjiang and Nanning. These results indicate a high diversity in the sweet potato viruses in southern China.
Coinfection of sweet potato in southern China
The co-infection rate was very high in our samples; most of the samples were infected by two or more virus species (Fig. 4). Compared to the 30.1% of the samples with a single virus, 17.4% were infected by two virus species, 10.5% had three virus species, and about 22.2% samples had four or more virus species. One sample was infected by seven species (Fig. 4).

Coinfection by sweet potato viruses in southern China. The number in the pie chart indicates the number of samples and percentage of each class
Geographic location of the sampling site was associated with differences in co-infecting viruses. In Nanning, co-infection was mainly due to three to four species of Potyvirus, while in Liuzhou and Meizhou, two to three species of Potyvirus coinfected samples. In the south part of southern China (Zhanjiang, Tunchang, Lingshui and Baoting) (Fig. 1), three species of Potyvirus always co-infected sweet potato. Guangzhou had the most severe co-infection situation; three to four species of Potyvirus plus two Begomovirus co-infected sweet potato there. Co-infection in Hezhou was not very serious, only about one-fourth samples had SPVG plus one another virus.
Discussion
Because southern China is one of the most important areas for sweet potato production in the world, understanding the incidence and distribution of viruses in the region is critical. We were able to use vsiRNA deep sequencing technology with traditional molecular detection methods to detect and confirm the incidence and distribution of 11 species of virus, including 5 species of Potyvirus and 2 of Begomovirus, from 219 samples from southern China.
Like other asexual crops, sweet potato accumulates many viruses during its growth and reproduction, and the co-infection by multiple viruses can significantly aggravate the disease symptoms and cause more serious yield losses. For example, SPFMV and SPCSV interact synergistically and cause greater yield reduction (Gutiérrez et al. 2003; Untiveros et al. 2007), although SPCSV was not found in our samples. However, in this study, it is difficult to elucidate the relationship between co-infection and symptoms in sweet potato. Of 219 field samples, which had a variety of symptoms, including healthy-looking samples, were detected to contain one or more viruses (Fig. 2). Because of the difference of sweet potato varieties, growth environment and infection stages, the same virus or multiple viruses showed different symptoms in different samples. In some samples, single virus infections showed severe symptoms, while multiple virus co-infections showed mild symptoms. The prevalence of viruses, especially two or more viruses in a sample, highlights the need for better strategies to prevent further spread of sweet potato viruses in most regions of southern China (Cuellar et al. 2015; Ngeve and Bouwkamp 1991). The only exception was Luoding, where only one virus species was detected, probably because there is less exchange of sweet potato varieties in this area and relatively fewer vector insects.
Recently, with the advent of high-throughput deep sequencing technology, deep sequencing of small RNAs has been widely applied for identifying viruses in eukaryotes (Chen et al. 2016; Kreuze et al. 2009, Su et al. 2015; Wu et al. 2010). In our study, although no new viruses were found in the samples, we obtained a general idea of the main viruses infecting sweet potato in southern China. Deep sequencing detected nine virus species (Table 1) in the one pool of 219 samples that were detected with greater frequency than other viruses when the 219 samples were individually tested by PCR/RT-PCR. This result suggests that these two methods are consistent and reliable for viruses in high titre. Not surprisingly, some viruses were detected only by PCR/RT-PCR, not by deep sequencing. Based on the reads number mapped to each virus by deep sequencing (Table 1), we conclude that the viruses detected only by PCR/RT-PCR may have been present in fewer samples or at lower titer. Therefore, combining the two methods will provide a more comprehensive standardized strategy for investigating viral diseases in the future.
Knowing that different Southern China regions can have different viruses or combinations of viruses should assist in developing management strategies to prevent further spread and keep plants virus-free. Such strategies should include the development of virus-free mother stock, removal of infected crops and control of insect vectors. Cultivation of virus-free plants is the most effective method to control sweet potato viruses, and efficient and economical multiple virus detection technology detection is a key in supporting this practice (Feng et al. 2000; Loebenstein 2015). Our investigation provides a basis for developing such technologies to support the prevention and control of sweet potato viruses in southern China, and with further development, deeps sequencing of vsiRNAs may become an efficient surveillance tool for sweet potato viruses.
References
Albuquerque LC, Inoue-Nagata AK, Pinheiro B, Resende RO, Moriones E, Navas-Castillo J (2012) Genetic diversity and recombination analysis of sweepoviruses from Brazil. Virol J 9:241
Chen S, Cao L, Huang Q, Qian Y, Zhou X (2016) The complete genome sequence of a novel maize-associated totivirus. Arch Virol 161:487–490
Clark CA, Davis JA, Abad JA, Cuellar WJ, Fuentes S, Kreuze JF, Gibson RW, Mukasa SB, Tugume AK, Tairo FD, Valkonen JPT (2012) Sweetpotato viruses: 15 years of progress on understanding and managing complex diseases. Plant Dis 96:168–185
Cuellar WJ, Galvez M, Fuentes S, Tugume J, Kreuze J (2015) Synergistic interactions of begomoviruses with sweet potato chlorotic stunt virus (genus Crinivirus) in sweet potato (Ipomoea batatas L.). Mol Plant Pathol 16:459–471
Feng G, Yifu G, Pinbo Z (2000) Production and deployment of virus-free sweetpotato in China. Crop Prot 19:105–111
Gibson R, Mwanga R, Kasule S, Mpembe I, Carey E (1997) Apparent absence of viruses in most symptomless field-grown sweet potato in Uganda. Ann Appl Biol 130:481–490
Gu YH, Tao X, Lai XJ, Wang HY, Zhang YZ (2014) Exploring the polyadenylated RNA virome of sweet potato through high-throughput sequencing. PLoS ONE 9:e98884
Gutiérrez D, Fuentes S, Salazar L (2003) Sweetpotato virus disease (SPVD): distribution, incidence, and effect on sweetpotato yield in Peru. Plant Dis 87:297–302
Hahn SK (1979) Effects of viruses (SPVD) on growth and yield of sweet potato. Exp Agric 15:253–256
Kreuze JF, Perez A, Untiveros M, Quispe D, Fuentes S, Barker I, Simon R (2009) Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virol 388:1–7
Loebenstein G (2012) Viruses in sweetpotato. Adv Virus Res 84:325–343
Loebenstein G (2015) Control of sweet potato virus diseases. Adv Virus Res 91:33–45
Luan YS, Zhang J, Liu DM, Li WL (2007) Molecular characterization of sweet potato leaf curl virus isolate from China (SPLCV-CN) and its phylogenetic relationship with other members of the Geminiviridae. Virus Genes 35:379–385
Moyer JW, Salazar L (1989) Viruses and virus-like diseases of sweet potato. Plant Dis 73:451–455
Ngeve JM, Bouwkamp JC (1991) Effects of sweet potato virus disease (SPVD) on the yield of sweet potato genotypes in Cameroon. Exp Agric 27:221–225
Su X, Fu S, Qian Y, Xu Y, Zhou X (2015) Identification of Hop stunt viroid infecting Citrus limon in China using small RNAs deep sequencing approach. Virol J 12:103
Untiveros M, Fuentes S, Salazar LF (2007) Synergistic interaction of Sweet potato chlorotic stunt virus (Crinivirus) with carla-, cucumo-, ipomo-, and potyviruses infecting sweet potato. Plant Dis 91:669–676
Wang Q, Zhang L, Wang B, Yin Z, Feng C, Wang Q (2010) Sweetpotato viruses in China. Crop Prot 29:110–114
Wu Q, Luo Y, Lu R, Lau N, Lai EC, Li W-X, Ding S-W (2010) Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc Natl Acad Sci USA 107:1606–1611
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Zhang L, Wang Q, Liu Q, Wang Q (2009) Sweetpotato in China. In: Loebenstein G, Thottappilly G (eds) The sweetpotato. Springer, New York, pp 325–358
Acknowledgements
This research was supported by the National Natural Science Foundation of China (grant no. 31601608) and the Science and Technology Planning Project of Guangdong Province (Grant no. 2017A020208058).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ma, S., Zheng, Q., Ye, J. et al. Identification of viruses infecting sweet potato in southern China by small RNA deep sequencing and PCR detection. J Gen Plant Pathol 85, 122–127 (2019). https://doi.org/10.1007/s10327-018-0832-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10327-018-0832-1