
Abstract
Deep sequencing of small RNA (sRNA) populations in maize plants from southwest China resulted in the identification of a previously unknown dsRNA virus with a sequence and genome organization resembling that of a totivirus. The complete viral genome is 3,956 nucleotides in length and contains two open reading frames (ORFs) with the potential to produce a ORF1-ORF2 fusion protein through a -1 ribosomal frameshift translation mechanism. ORF1 encodes the putative capsid protein (CP), whereas the predicted product of ORF2 contains motifs typical of an RNA-dependent RNA polymerase (RdRp). Phylogenetic analysis using the amino acid sequences of putative RdRp fusion proteins showed that the new virus was grouped in a clade together with the totiviruses, suggesting that it is a new member of the genus Totivirus of the family Totiviridae. The virus is tentatively named “maize-associated totivirus (MATV)”. Our findings demonstrate that it is feasible to identify totiviruses by deep sequencing of small RNAs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Members of the family Totiviridae have been found in a wide range of major fungal groups and include viruses from the genera Totivirus, Trichomonasvirus, Victorivirus, Giardiavirus and Leishmaniavirus. These viruses are characterized by double-stranded (ds) RNA genomes encapsidated in isometric virus particles with no lipid or carbohydrate content reported and no surface projections. The strategies employed for protein expression appear to differ among members of this family. Viruses in the genus Totivirus contain dsRNA genomes with two open reading frames. Typically, the 5’ ORF (ORF1) encodes a major coat protein (CP) and the 3’ ORF (ORF2) encodes a RNA-dependent RNA polymerase (RdRp) [1]. An RdRp fusion protein (ORF1-ORF2) is synthesized by the viral polymerase through a -1 ribosomal frameshift event [2, 3].
Conventional approaches of identifying viruses include DNA and RNA amplification by PCR and RT-PCR, respectively, serological detection, electron microscopy, and microarray hybridization [4], all of which require some prior knowledge of the agent(s) to be identified. Recently, with the advent of next-generation high-throughput parallel sequencing platforms, deep sequencing of small RNAs (sRNA) has been widely applied for identifying unknown viral agents in plants [5, 6]. Using this approach, Drosophila melanogaster totivirus (DTV), belonging to the family Totiviridae, was recently identified from Drosophila melanogaster cell lines by deep sequencing [7]. Here, a novel totivirus, provisionally named “maize-associated totivirus” (MATV), was identified in field-grown maize samples, and the complete genome sequence (3,956 nucleotides) of this virus was determined. Our findings demonstrate that deep sequencing of small RNAs is a feasible method for identification of totiviruses.
Five samples from symptomatic leaves of field-grown maize line Yahang 639 were collected in Dushan county, Guizhou province, China, in 2014. Total RNA was isolated from these maize leaves using TRIzol Reagent, following the manufacturer’s instructions (Invitrogen, Carlsbad, USA). The purified RNA preparation was used for construction of a small RNA library and sequencing. The resulting sRNA libraries were taken for further analysis using a custom bioinformatics pipeline. A total of 10,769,073 clean reads were generated after removing adaptor sequences and low-quality reads (Table S1). The population of reads in the 17- to 28-nt range were then compiled using an in-house Perl script for further assembly of contigs. A total of 619 contigs were generated using the Velvet program [8] with a k-mer value of 17 and were subsequently subjected to BLASTx analysis against the nonredundant (nr) database of the National Center for Biotechnology Information (NCBI) with standard parameters [7]. The comparisons demonstrated that 19 of the 619 contigs showed similarity to the RdRp-encoding sequence derived from totiviruses, with 50-82 % amino acid (aa) sequence identity, while six contigs closely matched the CP-encoding sequence from totiviruses, with 34-60 % aa sequence identity (Table S1). To confirm the presence of the putative novel virus, primers were designed based on the 25 identified contigs using the previously purified RNA as template. Two resulting fragments of 1309 bp and 2725 bp were obtained using the primer pairs F3 (5’-AGTGTTAAATATGAGTGGGCTAA-3’) + R3 (5’-TGTTCCTT GATCTAGAATCTAAT-3’) and F2 (5’-ACTACTCCAACATCCATGACA-3’) + R2 (5’-AGCCATCCGATGATGTATCTTA-3’), respectively. To determine the 5’- and 3’-terminal sequences of the complete genome sequence, 5’ and 3’ RACE-PCR was performed using a SMARTer™ RACE cDNA Amplification Kit (Clotech) according to the manufacturer’s instructions. This generated two fragments of 561 bp and 517 bp, respectively. The scheme for the cloning strategy of the full-length genome is shown in Fig. 1A. A full-length viral genome sequence of 3956 bp (GenBank accession number KP984504) was obtained by assembling the four fragments using the SeqMan software (Lasergene package, Version 7.1.0). A further RT-PCR analysis using primer pair F3/R3 and F2/R2 showed that two of the five maize samples, which displayed symptoms of leaf shrinkage, were infected by this novel maize-associated totivirus.

Analysis of the genome organization of MATV. (A) Schematic illustration of the cloning strategy used to obtain the full-length sequence of MATV. The relative positions of the primers used and sizes of the PCR products are shown (arrows and line segments, respectively). (B) Proposed genome organization of MATV. Boxes indicate the major ORFs predicted for the MATV genome. The name of the putative gene product is given above or below each box. Positions of the beginning and end of each ORF in the genome are labeled with Arabic numerals. The predicted frameshift heptanucleotide is indicated by a black triangle, and the ORF1-ORF2 fusion protein is indicted by a grey box. (C) Two necessary RNA elements for frameshifting. The putative slippery heptamer is indicated by a red box. The H-type pseudoknot is located immediately downstream of the putative slippery heptamer (color figure online)
The complete genome of this maize-associated totivirus had a G + C content of 51 %, and open reading frame analysis using the ORF finder function in the Snap Gene software [9] revealed the presence of two ORFs (ORF1 and ORF2), separated by 26 nt (Fig. 1B). ORF 1 (nt positions 114-1367) is predicted to encode a 417-aa protein, with the highest aa sequence identity (36 %) to the coat protein of Black raspberry virus F (BRVF, accession number YP_001497150.1), a putative member of the genus Totivirus. ORF2 (nt positions 1394-3898) is predicted to code for a 834-aa RNA-dependent RNA polymerase (RdRp) and contains the viral RdRp conserved domain RdRP_4 (accession number, pfam02123) [10]. The protein encoded by ORF2 exhibited the highest sequence identity (53 %) to the RdRp of BRVF (accession number YP_001497151.1).
Furthermore, an ORF1-ORF2 fusion protein is likely to be expressed via a -1 ribosomal frameshift translation strategy, like other members of the genus Totivirus [2, 3], producing a putative RdRp fusion protein with molecular mass of 139 kDa. The frameshift event is induced by two necessary RNA elements within the viral (+) mRNA: a) a potential slippery heptanucleotide sequence, X XXY YYZ (where X is A, C, G or U, Y is A or U, and Z is A, C or U) [11], and b) a downstream RNA pseudoknot [12]. In this case of the maize-associated totivirus, a heptanucleotide sequence, GGATTTT, located at nt 1256-1262 (Fig. 1B), was predicted to be a slippery sequence that could facilitate the -1 ribosomal frameshift, resulting in production of the ORF1-ORF2 fusion protein (Fig. 1B). Using the KnotSeeker program [13], an H-type pseudoknot, located immediately downstream of the putative slippery heptamer that could facilitate this phenomenon, was also detected at nt 1266-1298 (Fig. 1C).
Using amino acid sequences of RdRp fusion proteins from representative members of fungal and plant-infecting dsRNA viruses from the family Totiviridae, Chrysoviridae, Partitiviridae and Amalgaviridae (abbreviations and GenBank accession numbers are shown in Table S2), a phylogenetic tree was constructed using the neighbor-joining method in the MEGA 6.0 program, with a bootstrap test of 1000 replicates [14, 15]. Sequence comparisons and phylogenetic analysis clearly demonstrate that MATV groups perfectly with Ustilago maydis virus H1, Saccharomyces cerevisiae virus L-BC (La), Saccharomyces cerevisiae virus L-A, Ribes virus F (RVF) and BRVF (Fig. 2), which belong to the genus Totivirus, family Totiviridae. These members group separately from members of other selected families. It is worth noting that MATV formed a small cluster with totiviruses isolated from plants, such as RVF and BRVF (Fig. 2), suggesting that totiviruses originating from plants might be emerging within this genus.

Phylogenetic tree showing the relationship between the RdRP fusion proteins of MATV (blue) and other selected viruses. The tree was generated by the neighbor-joining method and is drawn to scale. Bootstrap values from 1000 replicates are indicated at each node, and only values greater than 50 % are shown (color figure online)
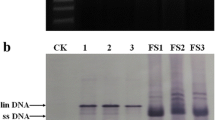
Totiviruses have been found in a wide range of major fungal groups. It is possible that MATV might have originated from indigenous or epiphytic fungi on maize plants. Hence, a PCR assay was carried out using the universal ITS primer pair ITS1/ITS4 to amplify fungal DNA. A fragment of about 690 bp was generated from the MATV-positive maize leaves and cloned into the pMD 18T-vector (Takara, Dalian, China) and sequenced (Fig. S1A). However, sequence similarity searches in the NCBI database indicated that the sequences of all 19 randomly selected clones were of plant origin (Fig. S1B), sharing 98-100 % nucleotide sequence identity with maize ribosomal RNA. Phylogenetic analysis showed that the above-mentioned 19 ITS sequences grouped with the ribosomal RNA sequences of different maize subspecies but were deeply separated from those derived from fungi that are hosts of the representative Totiviridae members (Fig. S1C), suggesting that MATV might be a novel maize-infecting virus. Subsequently, the entire dsRNA segment was extracted from MATV-infected maize leaves as described previously by DePaulo and Powell [16]. An approximately 4-kb dsRNA band was observed in gel electrophoresis analysis as expected (Fig. S2), suggesting the presence of the dsRNA virus genome in the field maize plants.
Collectively, the genome organization, sequence and phylogenetic analysis are consistent with the conclusion that MATV should be considered a member of a novel species in the genus Totivirus within the family Totiviridae. However, more work is necessary to demonstrate that the epidemiology of MATV represents a newly emerging field of investigation. Recent advances in deep sequencing have greatly accelerated the accuracy and rate of virus discovery. Our findings further demonstrate that the sRNAs deep sequencing approach is a feasible method for identification of totiviruses in field samples.
References
Knowles NJ, Hovi T, Hyypiä T, King AMQ, Lindberg AM, Pallansch MA, Zell R (2012) Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. Elsevier, Amsterdam, pp 855–880
Stielow B, Menzel W (2010) Complete nucleotide sequence of TaV1, a novel totivirus isolated from a black truffle ascocarp (Tuber aestivum Vittad). Arch Virol 155:2075–2078
Tzeng TH, Tu CL, Bruenn JA (1992) Ribosomal frameshifting requires a pseudoknot in the Saccharomyces cerevisiae double-stranded RNA virus. J Virol 66:999–1006
Grover V, Pierce ML, Melcher U (2007) Microarray hybridization for detection of plant viruses from natural settings. Phytopathology 97:S43–S43
Kreuze JF, Perez A, Untiveros M, Quispe D, Fuentes S, Barker I, Simon R (2009) Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology 388:1–7
Li RG, Gao S, Hernandez AG, Wechter WP, Fei ZJ, Ling KS (2012) Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS One 7:e37127
Wu QF, Luo YJ, Lu R, Lau N, Lai EC, Li WX, Ding SW (2010) Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc Natl Acad Sci 107:1606–1611
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Korf I (2004) Gene finding in novel genomes. BMC Bioinform 5:59
Bruenn JA (1993) A closely related group of RNA-dependent RNA polymerases from double-stranded RNA viruses. Nucleic Acids Res 21:5667–5669
Bekaert M, Rousset JP (2005) An extended signal involved in eukaryotic -1 frameshifting operates through modification of the E site tRNA. Mol Cell 17:61–68
Ten Dam EB, Pleij CWA, Bosch L (1990) RNA pseudoknots: translational frameshifting and readthrough on viral RNAs. Virus Genes 4:121–136
Sperschneider J, Datta A (2008) KnotSeeker: heuristic pseudoknot detection in long RNA sequences. RNA 14:630–640
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
DePaulo JJ, Powell CA (1995) Extraction of double-stranded RNA from plant tissues without the use of organic solvents. Plant Dis 79:246–248
Acknowledgments
This research was supported by a foundation for detecting plant virus using next-generation sequencing (grant 201310068) from General Administration of Quality Supervision, Inspection and Quarantine of China.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Chen, S., Cao, L., Huang, Q. et al. The complete genome sequence of a novel maize-associated totivirus. Arch Virol 161, 487–490 (2016). https://doi.org/10.1007/s00705-015-2657-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2657-y