
Abstract
Cytochrome P450 enzymes catalyse reactions of significant industrial interest but are underutilised in large-scale bioprocesses due to enzyme stability, cofactor requirements and the poor aqueous solubility and microbial toxicity of typical substrates and products. In this work, we investigate the potential for preparative-scale N-demethylation of the opium poppy alkaloid noscapine by a P450BM3 (CYP102A1) mutant enzyme in a whole-cell biotransformation system. We identify and address several common limitations of whole-cell P450 biotransformations using this model N-demethylation process. Mass transfer into Escherichia coli cells was found to be a major limitation of biotransformation rate and an alternative Gram-positive expression host Bacillus megaterium provided a 25-fold improvement in specific initial rate. Two methods were investigated to address poor substrate solubility. First, a biphasic biotransformation system was developed by systematic selection of potentially biocompatible solvents and in silico solubility modelling using Hansen solubility parameters. The best-performing biphasic system gave a 2.3-fold improvement in final product titre compared to a single-phase system but had slower initial rates of biotransformation due to low substrate concentration in the aqueous phase. The second strategy aimed to improve aqueous substrate solubility using cyclodextrin and hydrophilic polymers. This approach provided a fivefold improvement in initial biotransformation rate and allowed a sixfold increase in final product concentration. Enzyme stability and cell viability were identified as the next parameters requiring optimisation to improve productivity. The approaches used are also applicable to the development of other pharmaceutical P450-mediated biotransformations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cytochrome P450 enzymes (P450s) are promising enzymes for industrial and pharmaceutical biotechnology due to their ability to catalyse regioselective oxidations that are often difficult to achieve with synthetic organic chemistry [4]. While a vast array of useful P450 reactions have been identified, this enzyme family remains underutilised in industrial biocatalysis due to a range of typical process limitations [5, 38].
The relatively poor stability of many P450 enzymes, as well as their requirement for expensive NADPH cofactor, has led to the preference of whole-cell biotransformations over in vitro systems for preparative applications, as cells can provide both a protective environment for the enzyme and supply NADPH through metabolic regeneration [15]. While effective in this respect, whole-cell approaches have other limitations: the mass transfer of substrate into cells can be poor, and substrate and product toxicity can negatively affect the microbial host [47].
The rate of mass transfer of substrate across cell wall structures is often a significant limiting factor for biotransformations in whole-cell systems [11]. Both the rate and mechanism of mass transfer are strongly dependent on the physicochemical properties of the substrate (size, hydrophobicity, ionisation state) and the structural composition of the cell wall and membrane, which varies between organisms.
Substrates for P450s, particularly those for pharmaceutical applications, are often poorly water soluble, which is problematic for whole-cell biotransformations which take place in an aqueous environment. A common approach to tackle this issue is the use of biphasic biotransformation systems, which utilise an immiscible organic solvent phase in contact with the aqueous phase to act as a substrate reservoir [28]. Solvent selection is restricted, however, by the need for biocompatibility with the host organism, good substrate solubility in the solvent and favourable partitioning of the substrate into the aqueous phase. Other approaches to improve substrate loading focus on enhancing aqueous solubility, such as the use of miscible organic co-solvents, cyclodextrins [50] and recently the use of hydrophilic polymers [27].
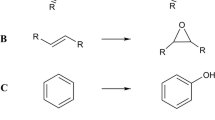
In a previous work [45], we screened a library of mutants of the bacterial P450 enzyme P450BM3 (CYP102A1) and identified a variant capable of selective (88%) N-demethylation of the opium poppy alkaloid noscapine, a drug with anticancer properties, at analytical scale (Fig. 1). This N-demethylated noscapine (N-nornoscapine) product is an essential precursor for N-modified noscapine analogues that have stronger antitumour activity [13, 35, 65] and have recently been shown to have antiparasitic activity [20]. In this work, we have investigated the potential for preparative-scale noscapine N-demethylation in a whole-cell biotransformation system using this P450BM3 mutant. We identify key factors limiting biotransformation and test several experimental approaches to address some of these limitations relating to mass transfer and substrate solubility.

Noscapine N-demethylation by P450BM3 mutant A3
Materials and methods
Chemicals, reagents and strains
Noscapine was provided by Sun Pharmaceutical Industries (Port Fairy, Australia). Molecular biology enzymes, nucleic acid purification kits and competent E. coli cells were purchased from New England Biolabs (Notting Hill, Australia). Primers for polymerase chain reactions (PCR) were synthesised by Integrated DNA Technologies (Singapore). Complex growth media components were Oxoid brand (Thermo Fisher Scientific, Scoresby, Australia). Lysozyme (chicken egg white) and achromopeptidase were purchased from Sigma-Aldrich (Castle Hill, Australia). Water used was of MilliQ grade (resistivity 18.2 mΩ). Methanol used for chromatographic separations was chromatography grade (Merck, Bayswater, Australia). Polyvinylpyrrolidone (average MW 40,000), (hydroxypropyl)methylcellulose (average Mn 86,000), (2-hydroxypropyl)-β-cyclodextrin (average Mw 1460) and all other chemicals were purchased from Sigma-Aldrich. Bacillus megaterium strain ATCC 14581™ was obtained from the American Type Culture Collection (Manassas, VA, USA). B. megaterium strain DSM-319 was obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ; Braunschweig, Germany). The plasmid pSPYngK-hp was a gift from Dieter Jahn (Addgene plasmid #48118).
Intracellular noscapine accumulation
Accumulation of cellular noscapine was measured using a method adapted from Williams and Piddock [62]. Glycerol stocks of B. megaterium ATCC 14589 and E. coli BL121(DE3) were used to inoculate shake flasks containing 50 mL and 120 mL of lysogeny broth (LB), respectively, which were shaken at 200 rpm and 30 °C for B. megaterium and 37 °C for E. coli for 14 h. Cells were collected by centrifugation at 3200×g and 10 °C for 20 min, washed twice with 2 × 10 mL potassium phosphate buffer (50 mM, pH 7.4) and resuspended in 1 mL of the same buffer. To measure noscapine uptake, a sample of 50 µL of concentrated cells was added to 200 µL of room-temperature phosphate buffer containing 100 µM noscapine (added from methanol stock; final MeOH concentration 1%). After the appropriate incubation time, 200 µL of this solution was rapidly cooled by mixing with 1 mL of phosphate buffer that was pre-chilled at − 4 °C in an ice/salt bath. The mixture was then immediately centrifuged at 20,000×g for 30 s at 4 °C. The supernatant was removed and the cell pellet was washed with a further 1 mL of pre-chilled buffer and centrifuged again. The wash buffer was completely removed and the pellet was stored in an ice/salt bath at − 4 °C until all samples were collected. To extract the accumulated noscapine, 100 µL of ice-cold methanol was added to each cell pellet and the pellets were incubated on ice for 20 min with brief vortexing every 5 min. The samples were then centrifuged at 18,000×g for 15 min at 4 °C to remove cell debris and then 30 µL of the supernatant was added to 70 µL of 0.1% trifluoroacetic acid (TFA) for analysis by ultra-high-performance liquid chromatography with a diode array detector (UHPLC-DAD) as previously described [45]. To estimate the amount of recovered noscapine that was adhered to the outside of cells rather than intracellularly located, the experiment was performed as above but the cell concentrate was added to noscapine/buffer solution that was pre-chilled at − 4 °C (a temperature at which no substrate is expected to cross the cell membrane [62]), incubated at − 4 °C for 3 min and then processed as before. Longer incubation times did not show any further increase in noscapine in the cell pellets. All noscapine accumulation experiments were performed in triplicate.
Plasmid assembly and transformation
A pET28a(+) vector (Merck, Australia) containing the gene for P450BM3 mutant A3 (R47L/A74G/F81I/F87A/L188Q/E267V) was constructed in previous work [45] and used here for recombinant expression in E. coli BL21(DE3). For expression of A3 in B. megaterium, the xylose-inducible expression vector p3STOP1632hp [51] was used as a backbone. The p3STOP1623hp backbone was obtained by PCR amplification of pSPYngK-hp [51] using a primer pair with sequences 5′-GGTACCGGCCGCATGCCG-3′ and 5′-TGTACATTCACCTCCTTGATTTAAGTGAACAAGTTTATCCATCAAC-3′. The A3 coding sequence was amplified from the pET28a (+) E. coli expression vector by PCR using a primer pair with sequences 5′-ATCAAGGAGGTGAATGTACAATGACAATTAAAGAAATGCCTCAGC-3′ and 5′-GCCGGCATGCGGCCGGTACCTTACCCAGCCCACACGTC-3′. The backbone and insert were assembled using the NEBuilder® HiFi DNA assembly kit (New England Biolabs) and transformed into chemically competent DH5-α cells for cloning. An empty p3STOP1632hp control vector was made by self-ligation of the amplified backbone using KLD Enzyme Mix (New England Biolabs). Protoplasts of B. megaterium strain DSM-319 were prepared and transformed according to Biedendieck et al. [6].
Noscapine solubility
For measurement of noscapine solubility in organic solvents, excess solid noscapine was added to 1 mL of solvent in glass vials and shaken at 200 rpm at ambient temperature for 24 h. The vials were then left for a further 48 h without shaking to allow sedimentation of undissolved noscapine. The saturated solvents were diluted into 100% methanol and noscapine was quantified by UHPLC-DAD, as previously described [45]. For solubility measurements in cyclodextrin and polymer solutions, 40 µL of a 100 mM noscapine stock in 5% tartaric acid was added to 1.96 mL of cyclodextrin and/or polymer in sodium/potassium phosphate buffer (100 mM, pH 6.75) to give a final noscapine concentration of 2 mM. Dilute tartaric acid was selected as a solvent for the noscapine stock due to its demonstrated ability to improve cyclodextrin solubilisation of weakly basic drugs [44]. The addition of the tartaric acid solution decreased the sample pH to 6.5. The solutions were equilibrated by shaking at 230 rpm at ambient temperature for 5 days, after which no change in noscapine solubility was found to occur. The solutions were filtered through a 0.45-µM nylon filter and diluted with 0.1% TFA before quantification by UHPLC-DAD.
Solubility modelling and in silico solvent screening
The solubility of noscapine in organic solvents was modelled by the modified Hansen parameter method developed by Louwerse et al. [33] using the associated MATLAB script written by the authors (provided in Supplementary Material for [33]). Empirical noscapine solubility measurements were made in 20 organic solvents, as described above, and used as inputs for the script program. Hansen parameters and molecular radii for solvents were taken from the HSPiP database (https://www.hansen-solubility.com/HSPiP/) and hydrogen bonding parameters were calculated manually, as described by Louwerse et al. [33]. The solubility measurements were provided as inputs to the program by grouping them into order-of-magnitude solubility intervals (i.e. 1–10 mM, 11–100 mM, etc.) and providing a ‘yes’ or ‘no’ term for each tested solvent in each interval. The program was then used to estimate Hansen solubility parameters for noscapine based on the empirical solubility measurements. These parameters were subsequently used as inputs for the program to predict noscapine solubility in a range of water-immiscible solvents with potential utility in biphasic biotransformation systems.
Whole-cell biotransformation
Cultures of E. coli BL21(DE3) containing the mutant P450BM3 enzyme A3 were prepared as described previously [45]. For protein expression in B. megaterium DSM-319, 50 mL of LB medium supplemented with 10 µg/mL tetracycline was inoculated with a glycerol stock of recombinant B. megaterium and shaken at 100 rpm and 37 °C overnight. A 1 mL volume of the overnight culture was used to inoculate 100 mL of Terrific Broth (TB) with 10 µg/mL tetracycline in a 500-mL shake flask and shaken at 230 rpm and 37 °C until the optical density at a wavelength of 600 nm (OD600) reached approximately 0.3. The culture was then cooled to 30 °C and expression was induced by the addition of 0.5% (w/v) xylose. Expression was continued for 20 h with 130 rpm shaking at 30 °C. Following expression, cells were collected by centrifugation at 3200×g and 10 °C for 20 min and the cell pellets were washed with 2 × 10 mL of nitrogen-free modified M9 medium (4.24 g/L Na2HPO4, 8.86 g/L KH2PO4, 2.5 g/L NaCl, 2 mM MgCl2, 0.1 mM CaCl2, 0.4% w/v glucose; pH 6.5). The cells were then resuspended in the same medium and diluted to give an OD600 of 5 for comparison of specific biotransformation rate with E. coli, or an OD600 of 35 for all other experiments. For biotransformation in the cyclodextrin/polymer solution, cells were resuspended in the same medium supplemented with 10% (w/v) (2-hydroxypropyl)-β-cyclodextrin (HP-β-CD) and 0.01% (w/v) hydroxypropyl methylcellulose (HPMC) and with the phosphate buffer components adjusted to give an initial pH of 6.65. Biotransformations were carried out in 2 mL volumes in 28-mL glass vials (LabTek, Brendale, Australia) at 230 rpm and 25 °C. Reactions were initiated by the addition of noscapine from a methanol stock (final MeOH concentration 1%) to give a noscapine concentration of 100 µM, or, for HP-β-CD/HPMC systems, by the addition of noscapine from a tartaric acid solution (final tartaric acid concentration 0.05%); the addition of tartaric acid lowered the pH to 6.5. For biphasic systems, a further addition of 0.5 mL of organic solvent saturated with noscapine was also made. At each sampling interval, 80 µL of aqueous phase and for biphasic systems, 20 µL of organic phase were removed and centrifuged at 16,000×g for 5 min. Cleared aqueous phase was diluted into a solution of 0.1% formic acid and 25% methanol and organic phase into 100% methanol for analysis by UPLC–UV–MS as below. For colony-forming unit (CFU) counts, samples were serially diluted in 0.85% saline and plated at 10−5, 10−6 and 10−7 dilutions onto LB agar and incubated at 37 °C. Glucose concentrations in the culture supernatant were measured spectroscopically using a glucose hexokinase (HK) assay kit (Sigma-Aldrich, Castle Hill, Australia). All biotransformation experiments were performed in triplicate.
Enzyme stability
To provide sufficient sample volumes for enzyme stability experiments, biotransformation reactions were performed essentially as above but scaled up to a volume of 13 mL and conducted in 250-mL shake flasks. One set of three replicates received 1% methanol without noscapine; one set was gently agitated at 60 rpm in round-bottom flasks; reactions were otherwise carried out in baffled flasks at 25 °C and shaken at 230 rpm with 100 µM noscapine added from methanol stock (final MeOH concentration 1%). At each sampling point, two 0.5 mL samples were removed for protein analysis and one 0.7 mL sample was removed for dry biomass measurement. The samples were centrifuged at 16,000×g for 5 min, the supernatant removed and pellets frozen at − 20 °C for biomass and SDS-PAGE analysis or − 80 °C for cytochrome P450 measurement. For P450 quantification, the cell pellets were thawed and washed with ice-cold potassium phosphate buffer (100 mM, pH 7.8), centrifuged and the supernatant removed. The pellets were resuspended in 50 µL of 2 mg/mL lysozyme and 0.2 mg/mL achromopeptidase in phosphate buffer (pH 7.8) and gently agitated for 45 min at room temperature. The suspensions were then diluted with 1 mL ice-cold potassium phosphate buffer (50 mM, pH 7.4), incubated on ice for 5 min and then sonicated (1.6 mm probe, 30% amplitude, 5 s on, 10 s off for six cycles) using a QSonica Q500 machine (QSonica LLC, Newtown, CT, USA). The lysate was then separated by centrifugation at 160,000×g for 40 min (Optima MAX™ ultracentrifuge; Beckman Coulter, Mount Waverley, Australia) and the supernatant was analysed for P450 content using carbon monoxide (CO)-difference spectroscopy according to the method of Omura and Sato [39]. No cytochrome P450 was detected by this method for control cells containing empty vector. For measurement of dry biomass weights, the cell pellets were resuspended in water, transferred to pre-weighed aluminium pans and dried in an oven for 24 h at 105 °C. Dried biomass was cooled in a vacuum desiccator over silica beads before weighing. Cell pellets for SDS-PAGE analysis were lysed according to the method of Barg et al. [3] and soluble protein fractions were run on a Bolt™ 4–12% Bis–Tris gel (Thermo Fisher Scientific, Scoresby, Australia). Gel loading volumes were adjusted to give an equal equivalent of dry biomass weight per lane and bands were visualised with Coomassie Brilliant Blue.
Substrate and product quantification
Quantification of substrate and product concentration was performed using an ACQUITY UPLC H-Class system equipped with PDA and QDa detectors. Analytes were separated with a Kinetix® biphenyl column (1.7 mm, 100 Å, 100 × 2.1 mm; Phenomenex, Lane Cove, Australia) maintained at 55 °C with a flow rate of 0.3 mL/min. A binary gradient method was used where the mobile phase consisted of (A) 0.1% formic acid and 10 mM ammonium formate in water and (B) 0.1% formic acid and 10 mM ammonium formate in methanol. The gradient elution program was as follows: isocratic at 25% solvent B for 30 s; 25 to 40% solvent B gradient over 30 s; 40 to 70% solvent B gradient over nine minutes; stepped to 95% solvent B and held for 1 min; then re-equilibration at 25% solvent B for 4 min. The QDa detector was operated in positive ion mode. Absorbance was monitored between 210 and 400 nm and a wavelength of 311 nm was used for substrate and product quantification. The products were identified by comparison of mass and retention time data with previous qTOF LC–MS/MS experiments [45].
Results and discussion
Microbial host selection
Substrate permeability
Our previous screening studies using cell lysates identified a P450BM3 pentamutant (R47L/A74G/F81I/F87A/L188Q), designated ‘A3’, capable of noscapine N-demethylation with good (88%) selectivity. To investigate the scale-up potential of this reaction, we aimed to incorporate the A3 mutant into a whole-cell biotransformation process for the N-demethylation of noscapine. As the mass transfer of substrate is often a major limitation in whole-cell drug biotransformations [11, 47], we examined whether there was any difference in noscapine uptake between Gram-negative and Gram-positive organisms prior to selecting a microbial host for the biotransformation bioprocess. These experiments were performed by incubating cell suspensions of either Escherichia coli or Bacillus megaterium with noscapine and quantifying intracellular noscapine accumulation at intervals up to 90 s; this timeframe was selected as a range of antibiotics have been shown to reach equilibrium in bacterial cells within similar time periods [1, 55, 62] and our preliminary experiments showed no significant further accumulation of noscapine after 90 s of incubation.
Noscapine accumulated rapidly in B. megaterium cells, reaching an equilibrium within 30–45 s, as shown in Fig. 2a. At the end of the assay, the accumulated noscapine was ~ 25-fold higher per unit biomass for B. megaterium cells than for E. coli cells (Fig. 2b). As it was likely that some of the noscapine measured in the cell pellets was adhered to the surface of the cells, rather than being located within the cells, we estimated the amount of adhered noscapine by performing extended incubations of cell suspensions with noscapine at – 4 °C. At this temperature, no diffusion across the cell membrane was expected to occur and all noscapine associated with the cell biomass was therefore considered to have adhered to the cell surface [62]. Under these conditions, the adhered noscapine was 0.30 ± 0.03 µg/mgdcw for B. megaterium and 0.05 ± 0.01 µg/mgdcw for E. coli. Notably, the concentration of noscapine that adhered to E. coli was similar to the total noscapine measured in the accumulation experiments in Fig. 2b, suggesting that only a small fraction of the noscapine associated with E. coli passed through the outer membrane of these cells. In contrast, noscapine that had adhered to B. megaterium represented only ~ 17% of the total noscapine measured in accumulation experiments after 90 s, indicating that most of the noscapine in the cell pellets had passed through the membrane of B. megaterium.

a Noscapine accumulation in Bacillus megaterium (filled square) vs Escherichia coli (filled circle). b Enlarged view of noscapine accumulation in E. coli. Error bars are ± 1 SD and are obscured by the symbol used as a marker for E. coli in (a)
These observed differences in noscapine uptake are consistent with differences in the uptake of drug-like substrates known to occur between Gram-positive and Gram-negative cells. Noscapine (pKa 6.3) is bulky, hydrophobic and was uncharged under the assay conditions used here (pH 7.4). Such substrates are expected to readily diffuse through the peptidoglycan and phospholipid bilayer of Gram-positive organisms but be inhibited by the dense, hydrophilic lipopolysaccharide coating of the Gram-negative outer membrane [37]. Consequently, Gram-positive organisms may be better hosts for whole-cell biotransformation of hydrophobic drugs, where mass transfer rates often limit productivity [11].
Whole-cell biotransformation
A B. megaterium expression system was developed to investigate whether the significant difference in substrate uptake between this organism and E. coli resulted in different rates of noscapine N-demethylation within a whole-cell biotransformation. The commonly used DSM-319 strain was transformed with a plasmid containing the A3 mutant P450BM3 gene under control of an optimised xylose-inducible promoter [51]. Biotransformations were performed using these cells in a resting state and compared with biotransformations performed using resting E. coli BL21(DE3) cells expressing the same enzyme under the IPTG-inducible T7 expression system.
Around ~ 25-fold more N-nornoscapine was produced per unit enzyme in the first 30 min using the B. megaterium system compared to E. coli, resulting in a ~ 12-fold higher specific yield after 4 h, as shown in Fig. 3. All noscapine turnover for both organisms was due to the mutant P450BM3 enzyme, as cells containing an empty vector showed no N-demethylation and no production of any other noscapine metabolite (results not shown). Both systems showed the same selectivity (~ 88%) for noscapine N-demethylation as previous cell lysate assays with this enzyme mutant (results not shown). The superior N-demethylation activity observed with B. megaterium compared to E. coli is presumably due to the greater permeability of B. megaterium cells to noscapine (Fig. 2). A similar observation was made by Bleif et al. [7] during the development of a whole-cell system for P450-mediated hydroxylation of the hydrophobic diterpene abietic acid, where biotransformation was successful using recombinant B. megaterium but no substrate conversion was detectable using E. coli despite the presence of a functional enzyme in these cells.

Noscapine N-demethylation by recombinant Bacillus megaterium (filled square) and Escherichia coli (filled circle) expressing P450BM3 mutant A3. Error bars are ± 1 SD and are obscured by the symbol used as a marker for E. coli
It is unlikely that the relatively poor performance of the E. coli system was due to a functional impairment of the mutant P450BM3 enzyme in this host. The concentration of cytochrome P450 enzyme in the biotransformation cultures was determined by CO-difference spectroscopy, which quantifies functional P450 enzyme [39], and the specific levels of mutant P450BM3 expression were comparable in the two different organisms (0.28 ± 0.02 nmol/mgdcw vs 0.45 ± 0.02 nmol/mgdcw for B. megaterium and E. coli, respectively). Furthermore, we have previously demonstrated the functional expression of this P450BM3 mutant in E. coli using cell lysates produced with the same expression vector and culture conditions used here [45].
While it is conceivable that the difference in biotransformation rate between the B. megaterium and E. coli systems is related to the different intracellular redox environments in the two hosts, this also seems unlikely, given that E. coli cells are reported to support far higher rates of NADPH-dependent biotransformation than the reaction rates observed here. Biotransformation studies using NADPH-dependent cyclohexanone monooxygenase, for example, have shown that glucose-fed, nitrogen-limited E. coli cells can maintain a significant excess of intracellular NADPH while sustaining a specific biotransformation rate of 1.1 µmol/(h mgdcw) [58, 59]. In the present work, conducted under similar conditions, the highest specific product formation rate of N-nornoscapine by E. coli cells (measured after 0.5 h of biotransformation) was 1.7 nmol/(h mgdcw), nearly 650-fold less than the previously described system. Even considering the differences in NADPH-to-product stoichiometry between the two enzymes (100% vs 16% coupling efficiency in vitro for cyclohexanone monooxygenase [14] and P450BM3 mutant A3 [45], respectively), it seems unlikely that the relatively poor performance of the E. coli system observed here was due to cofactor limitation in this host.
Selection of a host organism with good native permeability to a chosen substrate has several advantages over the alternative strategy of physically permeabilising cells following protein expression, e.g. by freeze–thawing or treatment with solvents or detergents [11]. Aside from requiring extra processing steps, the cellular damage caused by permeabilisation can be detrimental in cases where functional biocatalysis depends upon the active metabolism of the host, such as for the supply of NAD(P)H in P450-catalysed reactions. The use of cell permeabilisation to improve mass transfer is also limited to resting-cell biotransformations. The choice of B. megaterium as a host for noscapine biotransformation therefore avoids the mass transfer limitation encountered with E. coli while retaining process flexibility, as growing cells can be used if desired.
This work is one of several recent examples of using B. megaterium as a host for whole-cell biotransformations with recombinant P450 enzymes [7, 16, 23,24,25]. It is also, to the best of our knowledge, the first time that this organism has been used for a biotransformation with recombinant P450BM3 (CYP102A1), despite the enzyme being native to this species. Bacillus megaterium has broad potential as a biotransformation host due to its high capacity for heterologous protein expression and good plasmid stability [56]. We therefore chose to further investigate the potential of this host and enzyme system for preparative-scale N-demethylation of noscapine.
Increasing substrate supply
High concentrations of substrate are desirable in a biotransformation process to maximise volumetric productivity and reduce downstream processing requirements. Noscapine has poor aqueous solubility and low concentrations were used for the initial biotransformation experiments in Fig. 3 (100 µM or 41 mg/L added from a 10 mM methanol stock), as higher concentrations resulted in rapid precipitation. We therefore investigated two strategies to increase substrate supply in the biotransformation: a biphasic system and aqueous solubility enhancers.
Biphasic biotransformation
Solvent screening
A biphasic biotransformation system consists of an immiscible organic solvent that can supply a hydrophobic substrate to the aqueous phase containing the biocatalyst. The chosen solvent must be biocompatible with the host cell system, particularly where active host metabolism is required. The solvent should also provide good substrate solubility and partitioning between the aqueous and organic phases while also satisfying environmental, health and safety requirements. While the choice of potential solvents is vast, a systematic selection process can narrow down the number solvents to be tested experimentally to find an appropriate system [18].
We used a systematic screening strategy to find a suitable solvent based on published solvent properties and in silico solubility predictions made using Hansen solubility parameters [19]. A similar strategy was recently shown to be effective for the development of a biphasic system for geraniol biotransformation [42]. First, a solvent database (HSPiP; https://www.hansen-solubility.com/HSPiP/) was used to compile a list of solvents with octanol–water partition coefficient (log P) values greater than 3, as this measure provides an indication of potential biocompatibility [57]. This shortlist was then rationally narrowed to 15 solvents (Table 1) based on factors such as human and environmental safety, availability, cost, melting point and volatility. The shortlisted solvents were then assessed in silico for predicted noscapine solubility.
The Hansen solubility model predicts solvent–solute compatibility based on three parameters representing dispersion (δD), polar (δP) and hydrogen bonding (δH) interactions. Recently, Louwerse et al. [33] developed a modified Hansen model that incorporates several thermodynamic considerations for small molecule solutes and shows greater accuracy than the original model that was developed for solvent–polymer systems. The modified model and the associated MATLAB package were used to estimate Hansen parameters and predict noscapine solubility here. Noscapine solubility was initially measured in 20 readily available solvents (Table S1) and these data were used within the program for parameter estimation. A good fit (R2 = 0.88) was observed between the predicted and measured noscapine solubilities for the 20-solvent training dataset using the estimated noscapine Hansen parameters (δD = 28.7; δP= 11.3; δH = 7.8) (Fig. S1). These Hansen parameters were then applied to predict noscapine solubility within the shortlist for potential biphasic biotransformation solvents (Table 1).
Most of the solvents shortlisted for potential use in noscapine biotransformation were esters containing aromatic or medium-to-long-chain aliphatic groups. The compounds with the highest predicted noscapine solubilities (Table 1) contain either one or two aromatic functionalities; this appears to be due to the high Hansen dispersion parameter (δD) shared between these compounds and noscapine.
Three structurally diverse esters with good predicted noscapine solubility were selected for further investigation in a biotransformation experiment: butyl benzoate, heptyl acetate and dibutyl maleate. Isopropyl myristate was also included, as this compound has a significantly higher log P than the other selected solvents (8.5 vs 3.3–4.2). Whilst this solvent has a comparatively low predicted noscapine solubility (0.7 g/L), its inclusion allowed an investigation of a wider range of solvent hydrophobicity. Isopropyl myristate is also known to be effective in other biphasic biotransformations using hydrophobic small molecules [8, 30, 41, 61]. To our knowledge, the other three selected solvents have not previously been investigated for use with whole-cell biphasic systems, although dibutyl maleate was recently identified in a computational screen as a potentially suitable solvent for the biotransformation of flavour and fragrance compounds [41].
The measured solubility of noscapine in the four selected solvents is shown in Table 1. The measured solubilities were within 3 g/L of the predicted values, apart from butyl benzoate, for which the model gave a fourfold overestimation of noscapine solubility. These data not only demonstrate the potential utility of the modified Hansen model for in silico solvent screening of small drug molecules but also the need to validate predictions with experimental measurements.
Biphasic biotransformation
Isopropyl myristate was the most effective solvent for biphasic biotransformation, showing the highest yield of N-nornoscapine after 4 h compared to the use of a single aqueous phase or the three other solvents selected through in silico screening (Fig. 4). In each case, B. megaterium expressing the P450BM3 mutant A3 was used, with either a 20% volume of the selected solvent saturated with noscapine or a single-phase system containing 100 µM of noscapine with 1% methanol as a co-solvent. Approximately 2.3-fold more N-nornoscapine was produced using isopropyl myristate compared to the single-phase control, while the heptyl acetate and dibutyl maleate systems produced 6.3- and 8.8-fold less N-nornoscapine than the control, respectively. No substrate conversion was detected in the butyl benzoate solvent system.

Noscapine N-demethylation in a biphasic biotransformation system using the solvents isopropyl myristate (filled circle), dibutyl maleate (filled triangle), heptyl acetate (filled diamond) or butyl benzoate (inverted filled triangle). Biotransformation was also performed in a single aqueous phase as a control (filled square). Product concentrations are given as total product produced per volume of aqueous phase. Note that the marker for butyl benzoate (inverted filled triangle) appears partly obscured by the x axis, as biotransformation in this system was negligible. Error bars are ± 1 SD
The variation in product yields between the different solvent systems appeared to be primarily due to the toxicity of these solvents, as < 0.1% of cells remained viable in the biphasic systems containing heptyl acetate, dibutyl maleate and butyl benzoate following 4 h of biotransformation (Table 2). Butyl benzoate, despite having a log P between that of heptyl acetate and dibutyl maleate (Table 1), appeared to be particularly toxic as biotransformation activity was completely abolished (Fig. 4). This is likely related to the benzyl groups of this solvent, as alkylbenzenes have previously been shown to a have higher critical log P for biocompatibility than other solvent classes [57]. In contrast, cell viability in the isopropyl myristate system, which produced the highest yields, appeared similar to the viability observed for the single-phase control, with 11% and 14% of viable cells remaining for these reactions, respectively.
It is notable that B. megaterium cells containing an empty vector showed only a small viability loss under aqueous single-phase conditions (93% viable cells remaining; Table 2). The difference in survival between cells with the empty vector and cells containing the mutant P450BM3 enzyme in the single-phase control experiment (i.e. 93% vs 14% remaining viability) suggests that the expression of the enzyme lead to significant cellular toxicity. This reduction in cell viability may be due to the generation of reactive oxygen species by the P450 enzyme during non-productive catalytic cycles or toxicity associated with the N-nornoscapine product.
Retaining cell viability is important for the supply of NADPH cofactor to the enzyme during the biotransformation, and the retention of cell viability can also allow re-use of cells in multiple biotransformation cycles. While the general guideline is that solvents with log P > 4 are non-toxic to microbial cells, there are significant deviations from this rule of thumb depending on both the microbial strain and the molecular structure of the solvent [21, 28]. Organic solvents with log P between 3 and 5 are considered to have less predictable toxicities than those outside this range [57]. Based on the solvents screened here, B. megaterium requires higher solvent hydrophobicity for biocompatibility (i.e. isopropyl myristate); however, this comes at the cost of lower substrate loading (1.4 g/L in the organic phase), as noscapine solubility was highest in the more polar solvents that proved to be toxic (i.e. butyl benzoate and dibutyl maleate with 22.6 g/L and 19.6 g/L, respectively; Table 1).
The use of solvent-tolerant microbial strains is one approach to balance the need for good substrate solubility with biocompatibility [21]. It is important to consider, however, that some mechanisms of solvent resistance may also be detrimental to substrate uptake. For example, Gram-negative bacteria typically show higher solvent resistance than Gram-positive bacteria, attributed to the presence of an outer cell membrane. Our experiments suggest that the permeability barrier posed by the Gram-negative outer membrane is responsible for the low rate of noscapine biotransformation observed with E. coli (Fig. 3). While some particularly solvent-tolerant B. megaterium strains have been isolated [17, 43, 52], the properties conferring solvent resistance may impede substrate uptake. For example, a wild-type strain used for epoxide resolution in a biphasic system was active in the presence of solvents with log P > 3.2 [17] but displayed a low surface hydrophobicity and so may be less permeable to noscapine compared to the DSM-319 strain used here. Another important consideration is that many solvent resistance mechanisms in Gram-positive bacteria are adaptive responses [54]. Such responses are unlikely to occur with the nitrogen-limited, resting-cell biotransformation used here and improved tolerance may be seen with a growing-cell biotransformation process.
While the higher substrate loading in the isopropyl myristate system improved product titres compared to the single-phase system, there was an almost twofold decrease in the initial biotransformation rate (measured after 30 min of reaction). This was likely due to a lower aqueous concentration of substrate resulting from the partitioning of noscapine between the aqueous and organic phases. The aqueous phase in the isopropyl myristate system showed a steady, low noscapine concentration of around 30 µM throughout the biotransformation (data not shown). Maintaining a low aqueous concentration is advantageous when a biphasic system is employed due to substrate toxicity, but in this case, our objective was to overcome poor substrate solubility and so a decrease in reaction rate due to the limited aqueous substrate concentration is a potential disadvantage. We therefore investigated an alternative strategy of improving aqueous noscapine solubility to drive faster reaction rates.
Improving aqueous solubility
Cyclodextrins are often employed as pharmaceutical excipients due to their ability to enhance drug solubility. This class of compounds was investigated here to assess whether the aqueous solubility of noscapine could be improved. Cyclodextrins form soluble inclusion complexes with drugs and have previously been used to improve whole-cell biotransformation rates of hydrophobic compounds such as steroids [36, 49, 60]. Cyclodextrins have also been used to enhance biotransformation by recombinant B. megaterium [23]. In some pharmaceutical applications, small amounts of water-soluble polymers such as polyvinylpyrrolidone (PVP) and hydroxypropyl methylcellulose (HPMC) have also been shown to improve drug–cyclodextrin complexation and allow further enhancement of drug solubility [31], though these ternary complexes have not previously been employed in whole-cell biotransformations. The cyclodextrin (2-hydroxypropyl)-β-cyclodextrin (HP-β-CD), a widely used and highly soluble cyclodextrin derivative, as well as the polymers PVP and HPMC, was investigated here for their effects on noscapine solubility.
The combination of 10% HP-β-CD with 0.1% of polymer HPMC provided a significant increase in aqueous noscapine solubility (Fig. 5), allowing a ~ 16-fold enhancement in noscapine concentration to 835 µM (345 mg/L) compared to the 50 µM of noscapine dissolved in the control solution containing only phosphate buffer. Solutions of 5% and 10% HP-β-CD alone gave more modest three- and fourfold enhancements in noscapine solubility, respectively, and a 0.1% solution of HPMC polymer provided a threefold enhancement, indicating that HP-β-CD and HPMC act synergistically to enhance noscapine solubility. This effect was still apparent when a lower polymer concentration of 0.01% HPMC was used with 10% HP-β-CD, which gave a 14.5-fold enhancement in noscapine concentration (736 µM; 304 mg/L) with considerably less viscosity compared to solutions containing 0.1% HPMC polymer. In contrast, a solution of 0.1% PVP had negligible effect on the solubility of noscapine with or without HP-β-CD addition.

Aqueous solubility of noscapine in solutions of cyclodextrin, polymer or combinations of cyclodextrin and polymer. Abbreviations: HP-β-CD, (2-hydroxypropyl)-β-cyclodextrin; PVP, polyvinylpyrrolidone; HPMC, (hydroxypropyl)methylcellulose. Solutions were prepared in phosphate buffer (100 mM) at pH 6.5. Error bars are ± 1 SD
The combination of cyclodextrin HP-β-CD and polymer HPMC not only provided significant enhancements in noscapine solubility but also functioned well as a precipitation inhibitor [63] for supersaturated noscapine solutions. When measuring the equilibrium solubility of noscapine in HP-β-CD/polymer systems, we observed that the HP-β-CD/HPMC systems remained optically clear for extended periods following addition of excess noscapine, whereas precipitate was visible in all other cyclodextrin or polymer solutions within 1 h. Under the same conditions of temperature and agitation as the biotransformation experiments, a solution of 10% HP-β-CD with 0.01% HPMC could maintain a noscapine concentration of ~ 1 mM for at least 24 h (Fig. S2). We therefore investigated biotransformation performance in solutions of 10% HP-β-CD with 0.01% HPMC using noscapine concentrations up to 1 mM.
The higher concentrations of noscapine enabled by HP-β-CD/HPMC addition led to increased biotransformation productivity, as shown for experiments performed using an initial substrate concentration of 100 µM, 500 µM or 1 mM (Fig. 6). The initial rate measured at 30 min was found to increase up to eightfold between the reactions with 100 µM and 1 mM noscapine, allowing a final product concentration up to 266 µM (110 mg/L). This product titre was a 6.4-fold improvement over the single-phase system in 1% methanol (Fig. 4) where noscapine was used at its maximum solubility of 100 µM. This product titre was also 2.8-fold higher than the best-performing biphasic system (IPM; Fig. 4).

Noscapine N-demethylation by biotransformation in solutions of 10% (2-hydroxypropyl)-β-cyclodextrin and 0.01% (hydroxypropyl)methylcellulose with a starting substrate concentration of (filled square) 100 µM, (filled circle) 500 µM or (filled triangle) 1 mM noscapine. Error bars are ± 1 SD
While the aqueous solubility enhancers allowed significantly higher final yield (266 µM vs 41 µM), the initial rate of N-demethylation with 100 µM noscapine and HP-β-CD/HPMC (Fig. 6) was slower than in the equivalent reaction with 100 µM noscapine without HP-β-CD/HPMC (Fig. 4), with 1.6-fold less product at 30 min, though both systems eventually reached the same final yield. This may be because the complexation of noscapine with cyclodextrin resulted in a lower concentration of free substrate. Alternatively it may be due to decreased mass transfer from the higher viscosity of the HP-β-CD/HPMC solution [32]. A similar observation was made by Kiss et al. [23] for a B. megaterium biotransformation system using 5% HP-β-CD, which showed slower initial reaction rates with cyclodextrin compared to a control reaction with 5% DMSO in shake flask experiments. When the reaction was performed in bioreactors, however, the HP-β-CD system showed faster rates than the control, suggesting that stronger mixing may improve the initial rates in HP-β-CD/HPMC here.
While allowing higher final product titres, the HP-β-CD/HPMC solutions also showed some toxicity towards B. megaterium with only 2–3% of initial cell viability remaining after 4 h, whereas 14% of cells remained viable in the control biotransformation (100 µM noscapine, 1% methanol; Table 2). This difference appeared to be entirely due to the solubility enhancers as the remaining viability in the HP-β-CD/HPMC systems was the same for both 100 µM and 1 mM noscapine, indicating no additional toxicity from higher concentrations of substrate or product. While often considered to have good biocompatibility [2], cyclodextrins and their derivatives have been shown to be toxic to microbial cells in some cases. This appears to be highly dependent on both the microbial species and variant of cyclodextrin [48, 67, 68]. At 15% concentration, HP-β-CD has been shown to cause membrane damage and protein leakage to Gram-positive Arthrobacter simplex cells [48]; this is also likely responsible for the observed toxicity here. It is unlikely that the low concentration (0.01% w/v) of HPMC exerted any toxicity as this polymer is often used at higher concentrations in dietary probiotic formulations without loss of cell viability [40, 64].
Cofactor/viability limitation
While the higher substrate concentrations achieved by the addition of HP-β-CD/HPMC significantly improved initial reaction rates, the experiments indicated that another factor such as cofactor supply, cell viability or enzyme stability became limiting early in the biotransformation. After 1 h of biotransformation, product formation rates in the reactions containing high and moderate concentrations of noscapine (1 mM and 500 µM, Fig. 6) were similar, which is not in keeping with the first-order reaction kinetics expected in this substrate range. This behaviour lead us to examine cofactor supply and cell viability as potential limiting factors.
The sustained supply of NADPH to the reaction requires cells to have access to a carbon source and to be metabolically active. To investigate whether glucose was actively consumed by the host cells throughout the biotransformation and did not become exhausted, we monitored the glucose consumption and product formation in a biotransformation with 1 mM noscapine in a HP-β-CD/HPMC solution (Fig. 7). Product formation was also monitored in the absence of glucose, where regeneration of NADPH is expected to be limited.

Noscapine N-demethylation with (filled circle) and without (filled square) glucose added to the biotransformation. Glucose concentration is also shown for the relevant biotransformation (open square). Error bars are ± 1 SD
A concentration of 22 mM (0.4% w/v) glucose was found to be more than sufficient to supply the cells over a 4 h period, as shown in Fig. 7. The presence of glucose resulted in 1.4-fold more product after 1 h, likely due to higher NADPH supply. At later timepoints, however, the rate of product formation was similar in the presence and absence of glucose, with an increase of ~ 50 µM between 1 and 4 h (Fig. 7), despite active glucose consumption in the glucose-fed culture and an order of magnitude difference in cell viability measured after 4 h (2% vs 0.1% for cells with and without glucose, respectively; Table 2). These differences indicate that neither glucose supply nor cell viability were the key factor limiting biotransformation rate after 1 h. This suggested that the observed decline in product formation may be have been due to the loss of active P450 enzyme.
Enzyme stability
The stability of the mutant P450BM3 enzyme was assessed during biotransformation to determine whether this factor limits the productivity of the system and could be the focus of future optimisation studies. The concentration of cytochrome P450 enzyme in cell pellets collected from a standard biotransformation was measured using carbon monoxide (CO)-difference spectroscopy [39] and the concentration of soluble intracellular protein visualised using SDS-PAGE. We also investigated the potential effects of dissolved oxygen and substrate turnover on enzyme stability using reactions performed with low agitation or in the absence of substrate.
A significant loss of P450 enzyme occurred over the course of the biotransformation reactions performed under conditions of high agitation, as assessed by CO-difference spectroscopy (Fig. 8a). The cytochrome P450 content of the cells dropped by four- to fivefold over 4 h, while the weight of dry biomass and the concentration of soluble P450BM3 protein visualised by SDS-PAGE remained relatively constant (Fig. 7b). This observation likely explains the decline in product formation seen after 1 h of biotransformation in Fig. 6, where high agitation was used. As the spectroscopic method quantifies P450-bound haem, it seems likely that enzyme deactivation was due to haem loss or damage rather than proteolysis or thermal denaturation and precipitation of the P450 enzyme.

a Concentration of active cytochrome P450 enzyme per unit biomass in a biotransformation measured by carbon monoxide-difference spectroscopy performed with (filled circle) high agitation and 100 µM substrate; (filled square) high agitation without substrate; or (filled triangle) low agitation and 100 µM substrate. The corresponding dry biomass weight for each time point are also shown (open square). Error bars are ± 1 SD. b SDS-PAGE gel showing the P450BM3 enzyme polypeptide band at 121 kDa, indicated by an arrow. Lanes are representative samples from experiments in part (a). Lane 1, before biotransformation; lanes 2–4, following 4 h of biotransformation performed at: high agitation and 100 µM substrate (lane 2); (filled square) high agitation without substrate (lane 3); or low agitation and 100 µM substrate (lane 4). A sample taken from cells containing an empty vector is also shown (c)
A similar loss of P450 enzyme occurred both with and without substrate addition, indicating that enzyme deactivation was related to the conditions of high agitation rather than substrate turnover. In contrast, only ~ 10% of cytochrome P450 enzyme was lost in the reaction performed with gentle agitation, though subsequent biotransformation experiments using low agitation showed significantly less productivity (2.8-fold less product after 4 h at 70 rpm compared to 230 rpm; 1 mM substrate in HP-β-CD/HPMC; Fig. S3), likely due to a lower dissolved oxygen concentration under these conditions.
The cytochrome P450 haem group can be damaged by hydrogen peroxide and other reactive oxygen species (ROS) produced by the reduction of molecular oxygen during non-productive P450 catalytic cycles [22]. This can occur when product formation is poorly coupled to NADPH reduction or when no substrate is present [66]. Given that the P450BM3 mutant used in this work only showed only 16% coupling efficiency in vitro [45] and its stability was improved under low aeration (Fig. 8a), this mechanism may be responsible for the loss of activity observed during biotransformation. Production of ROS by the recombinant P450BM3 enzyme may also contribute to the loss of cell viability observed during biotransformation experiments (a reduction in cell viability from 93% to 14% was observed after 4 h of reaction with P450-expressing cells compared to cells with an empty vector; Table 2). Our observations are consistent with the poor stability of other P450 enzymes which has previously been shown to be a significant factor limiting whole-cell biotransformation productivity [23, 34].
One strategy to improve both enzyme stability and cell survival would be to co-express ROS scavengers [12, 29], such as catalase or superoxide dismutase, together with the recombinant P450BM3 mutant. This approach would also need to consider the net benefit to biotransformation productivity, however, as co-expression strategies may result in reduced P450BM3 expression [46]. Additionally, these ROS scavengers are natively expressed in aerobic bacteria to mitigate oxidative stress [10], so it unclear whether the overexpression of these enzymes would have any further protective effects on the enzyme or host cell under these conditions.
Another strategy could be to change the mode of operation from a resting-cell biotransformation to a growing-cell process. All biotransformations performed in this work used a nitrogen-free medium, which does not allow for further protein expression during the biotransformation or significant adaptive cell responses to environmental challenges. Performing the noscapine biotransformation concurrently with recombinant P450 expression and cellular growth may allow for improved product titres, as the unstable enzyme would be continuously replaced and actively growing cells may be better able to respond to ROS generation. Growing cells may also be more tolerant to organic solvents and cyclodextrins than resting cells. A common disadvantage of growing-cell processes, however, is the reduced availability of cellular cofactors, such as NADPH, due to the energetic demands of biomass formation [9]. This can be further addressed using cofactor recycling or metabolic engineering strategies [26].
These strategies, together with those already successfully employed in this study, are presented in Table 3. This table summarises the productivity and yield gains made using alternative hosts, biphasic systems and solubility enhancers. Whilst the highest volumetric productivity achieved is one to two orders of magnitude below most reported industrial pharmaceutical biotransformations [53], the process limitations encountered in this work are typical of P450-mediated drug biotransformations. Further consideration of the factors limiting biotransformation and the strategies identified for optimisation will potentially allow improvements in enzyme activity, cofactor supply and cell viability to enhance biotransformation productivity for the N-demethylation of noscapine.
Conclusions
We have incorporated a drug-metabolising P450BM3 enzyme mutant into a whole-cell biotransformation process, showing proof of concept for preparative-scale biocatalytic N-demethylation of noscapine. Several factors limiting productivity were identified and strategies to increase productivity systematically assessed. The highest productivity of 27.5 mg/(L h)) could be achieved by employing a B. megaterium host to overcome mass transfer limitations and cyclodextrins and polymers to enhance the aqueous solubility of noscapine. Biphasic systems developed using in silico solubility modelling could also be used to increase productivity (to 9.8 mg/(L h)). Strategies that can improve the initial rates of biotransformation, such as the use of aqueous solubility enhancers, were shown to be particularly important for this system as the mutant P450BM3 enzyme was found to have low stability under whole-cell biotransformation conditions. Maintenance of cell viability was also identified as a significant challenge.
This study shows that significant improvements in productivity and yield can be obtained by relatively simple changes in biotransformation parameters using a systematic debottlenecking approach. The strategies used in this work can also be applied to examine and optimise other whole-cell P450-based systems.
References
Asuquo AE, Piddock LJV (1993) Accumulation and killing kinetics of fifteen quinolones for Escherichia coil, Staphylococcus aureus and Pseudomonas aeruginosa. J Antimicrob Chemother 31:865–880
Bar R (1989) Cyclodextrin-aided bioconversions and fermentations. Trends Biotechnol 7:2–4
Barg H, Malten M, Jahn M, Jahn D (2005) Protein and vitamin production in Bacillus megaterium. In: Barredo J (ed) Microbial processes and products. Humana Press, Totowa, pp 205–223
Bernhardt R (2006) Cytochromes P450 as versatile biocatalysts. J Biotechnol 124:128–145
Bernhardt R, Urlacher VB (2014) Cytochromes P450 as promising catalysts for biotechnological application: chances and limitations. Appl Microbiol Biotechnol 98:6185–6203. https://doi.org/10.1007/s00253-014-5767-7
Biedendieck R, Borgmeier C, Bunk B, Stammen S, Scherling C, Meinhardt F, Wittmann C, Jahn D (2011) Systems biology of recombinant protein production using Bacillus megaterium. In: Jameson D, Verma M, Westerhof HV (eds) Methods in enzymology, vol 500. Academic Press, Cambridge, pp 165–195
Bleif S, Hannemann F, Lisurek M, von Kries JP, Zapp J, Dietzen M, Antes I, Bernhardt R (2011) Identification of CYP106A2 as a regioselective allylic bacterial diterpene hydroxylase. ChemBioChem 12:576–582. https://doi.org/10.1002/cbic.201000404
Brennan TCR, Turner CD, Krömer JO, Nielsen LK (2012) Alleviating monoterpene toxicity using a two-phase extractive fermentation for the bioproduction of jet fuel mixtures in Saccharomyces cerevisiae. Biotechnol Bioeng 109:2513–2522
Buhler B, Park JB, Blank LM, Schmid A (2008) NADH availability limits asymmetric biocatalytic epoxidation in a growing recombinant Escherichia coli strain. Appl Environ Microbiol 74:1436–1446. https://doi.org/10.1128/AEM.02234-07
Cabiscol E, Tamarit J, Ros J (2000) Oxidative stress in bacteria and protein damage by reactive oxygen species. Int Microbiol 3(1):3–8
Chen RR (2007) Permeability issues in whole-cell bioprocesses and cellular membrane engineering. Appl Microbiol Biotechnol 74:730–738
Das S, Glenn JH, Subramanian M (2010) Enantioselective oxidation of 2-hydroxy carboxylic acids by glycolate oxidase and catalase coexpressed in methylotrophic Pichia pastoris. Biotechnol Prog 26:607–615. https://doi.org/10.1002/btpr.363
DeBono AJ, Xie JH, Ventura S, Pouton CW, Capuano B, Scammells PJ (2012) Synthesis and biological evaluation of N-substituted noscapine analogues. ChemMedChem 7:2122–2133. https://doi.org/10.1002/cmdc.201200365
Donoghue NA, Norris DB, Trudgill PW (1976) The purification and properties of cyclohexanone oxygenase from Nocardia globerula CL1 and Acinetobacter NCIB 9871. Eur J Biochem 63:175–192
Duetz WA, van Beilen JB, Witholt B (2001) Using proteins in their natural environment: potential and limitations of microbial whole-cell hydroxylations in applied biocatalysis. Curr Opin Biotechnol 12:419–425. https://doi.org/10.1016/S0958-1669(00)00237-8
Gerber A, Milhim M, Hartz P, Zapp J, Bernhardt R (2016) Genetic engineering of Bacillus megaterium for high-yield production of the major teleost progestogens 17α, 20β-di- and 17α, 20β, 21α-trihydroxy-4-pregnen-3-one. Metab Eng 36:19–27. https://doi.org/10.1016/j.ymben.2016.02.010
Gong PF, Xu JH (2005) Bio-resolution of a chiral epoxide using whole cells of Bacillus megaterium ECU1001 in a biphasic system. Enzyme Microb Technol 36:252–257. https://doi.org/10.1016/j.enzmictec.2004.07.014
Grundtvig IPR, Heintz S, Kruhne U, Gernaey KV, Adlercreutz P, Hayler JD, Wells AS, Woodley JM (2018) Screening of organic solvents for bioprocesses using aqueous-organic two-phase systems. Biotechnol Adv 36:1801–1814. https://doi.org/10.1016/j.biotechadv.2018.05.007
Hansen CM (1967) The three dimensional solubility parameter. Danish Technical, Copenhagen, p 14
Harikandei KB, Salehi P, Ebrahimi SN, Bararjanian M, Kaiser M, Khavasi HR, Al-Harrasi A (2019) N-substituted noscapine derivatives as new antiprotozoal agents: synthesis, antiparasitic activity and molecular docking study. Bioorg Chem 91:103116
Heipieper HJ, Neumann G, Cornelissen S, Meinhardt F (2007) Solvent-tolerant bacteria for biotransformations in two-phase fermentation systems. Appl Microbiol Biotechnol 74:961–973. https://doi.org/10.1007/s00253-006-0833-4
Karuzina II, Zgoda VG, Kuznetsova GP, Samenkova NF, Archakov AI (1999) Heme and apoprotein modification of cytochrome P450 2B4 during its oxidative inactivation in monooxygenase reconstituted system. Free Radic Biol Med 26:620–632. https://doi.org/10.1016/s0891-5849(98)00252-4
Kiss FM, Lundemo MT, Zapp J, Woodley JM, Bernhardt R (2015) Process development for the production of 15β-hydroxycyproterone acetate using Bacillus megaterium expressing CYP106A2 as whole-cell biocatalyst. Microb Cell Fact 14:28. https://doi.org/10.1186/s12934-015-0210-z
Kleser M, Hannemann F, Hutter M, Zapp J, Bernhardt R (2012) CYP105A1 mediated 3-hydroxylation of glimepiride and glibenclamide using a recombinant Bacillus megaterium whole-cell catalyst. J Biotechnol 157:405–412. https://doi.org/10.1016/j.jbiotec.2011.12.006
König L, Hartz P, Bernhardt R, Hannemann F (2019) High-yield C11-oxidation of hydrocortisone by establishment of an efficient whole-cell system in Bacillus megaterium. Metab Eng 55:59–67
Lee WH, Kim MD, Jin YS, Seo JH (2013) Engineering of NADPH regenerators in Escherichia coli for enhanced biotransformation. Appl Microbiol Biotechnol 97:2761–2772. https://doi.org/10.1007/s00253-013-4750-z
Lee PG, Lee SH, Kim J, Kim EJ, Choi KY, Kim BG (2018) Polymeric solvent engineering for gram/liter scale production of a water-insoluble isoflavone derivative, (S)-equol. Appl Microbiol Biotechnol 102:6915–6921. https://doi.org/10.1007/s00253-018-9137-8
Leon R, Fernandes P, Pinheiro H, Cabral J (1998) Whole-cell biocatalysis in organic media. Enzyme Microb Technol 23:483–500
Liu W, Xu X, Zhang R, Cheng T, Cao Y, Li X, Guo J, Liu H, Xian M (2016) Engineering Escherichia coli for high-yield geraniol production with biotransformation of geranyl acetate to geraniol under fed-batch culture. Biotechnol Biofuels 9:58
Liu Q, Ma X, Cheng H, Xu N, Liu J, Ma Y (2017) Co-expression of l-glutamate oxidase and catalase in Escherichia coli to produce alpha-ketoglutaric acid by whole-cell biocatalyst. Biotechnol Lett 39:913–919. https://doi.org/10.1007/s10529-017-2314-5
Loftsson T, Másson M (2004) The effects of water-soluble polymers on cyclodextrins and cyclodextrin solubilization of drugs. J Drug Deliv Sci Technol 14:35–43
Loftsson T, Frikdriksdóttir H, Sigurkdardóttir AM, Ueda H (1994) The effect of water-soluble polymers on drug-cyclodextrin complexation. Int J Pharm 110:169–177
Louwerse MJ, Maldonado A, Rousseau S, Moreau-Masselon C, Roux B, Rothenberg G (2017) Revisiting hansen solubility parameters by including thermodynamics. ChemPhysChem 18:2999–3006. https://doi.org/10.1002/cphc.201700408
Lundemo MT, Notonier S, Striedner G, Hauer B, Woodley JM (2016) Process limitations of a whole-cell P450 catalyzed reaction using a CYP153A-CPR fusion construct expressed in Escherichia coli. Appl Microbiol Biotechnol 100:1197–1208. https://doi.org/10.1007/s00253-015-6999-x
Manchukonda NK, Naik PK, Santoshi S, Lopus M, Joseph S, Sridhar B, Kantevari S (2013) Rational design, synthesis, and biological evaluation of third generation α-noscapine analogues as potent tubulin binding anti-cancer agents. PloS One 8(10):e77970. https://doi.org/10.1371/journal.pone.0077970
Manosroi A, Saowakhon S, Manosroi J (2008) Enhancement of androstadienedione production from progesterone by biotransformation using the hydroxypropyl-beta-cyclodextrin complexation technique. J Steroid Biochem Mol Biol 108:132–136. https://doi.org/10.1016/j.jsbmb.2007.05.032
Nikaido H (1994) Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science 264:382–388
O’Reilly E, Kohler V, Flitsch SL, Turner NJ (2011) Cytochromes P450 as useful biocatalysts: addressing the limitations. Chem Commun (Camb) 47:2490–2501. https://doi.org/10.1039/c0cc03165h
Omura T, Sato R (1964) The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem 239:2370–2378
Pop OL, Brandau T, Schwinn J, Vodnar DC, Socaciu C (2015) The influence of different polymers on viability of Bifidobacterium lactis 300b during encapsulation, freeze-drying and storage. J Food Sci Technol 52:4146–4155. https://doi.org/10.1007/s13197-014-1441-4
Priebe X, Daugulis AJ (2018) Thermodynamic affinity-based considerations for the rational selection of biphasic systems for microbial flavor and fragrance production. J Chem Technol Biot 93:656–666. https://doi.org/10.1002/jctb.5524
Priebe X, Daschner M, Schwab W, Weuster-Botz D (2018) Rational selection of biphasic reaction systems for geranyl glucoside production by Escherichia coli whole-cell biocatalysts. Enzyme Microb Technol 112:79–87. https://doi.org/10.1016/j.enzmictec.2017.11.003
Priya JDA, Divakar K, Prabha MS, Selvam GP, Gautam P (2014) Isolation, purification and characterisation of an organic solvent-tolerant Ca2+-dependent protease from Bacillus megaterium AU02. Appl Biochem Biotechnol 172:910–932
Redenti E, Szente L, Szejtli J (2000) Drug/cyclodextrin/hydroxy acid multicomponent systems. Properties and pharmaceutical applications. J Pharm Sci 89:1–8
Richards L, Lutz A, Chalmers DK, Jarrold A, Bowser T, Stevens GW, Gras SL (2019) Production of metabolites of the anti-cancer drug noscapine using a P450BM3 mutant library. Biotechnol Rep (Amst) 24:e00372. https://doi.org/10.1016/j.btre.2019.e00372
Schewe H, Holtmann D, Schrader J (2009) P450 BM-3-catalyzed whole-cell biotransformation of α-pinene with recombinant Escherichia coli in an aqueous–organic two-phase system. Appl Microbiol Biotechnol 83:849–857
Schrewe M, Julsing MK, Buhler B, Schmid A (2013) Whole-cell biocatalysis for selective and productive C–O functional group introduction and modification. Chem Soc Rev 42:6346–6377. https://doi.org/10.1039/c3cs60011d
Shen Y, Wang M, Zhang L, Ma Y, Ma B, Zheng Y, Liu H, Luo J (2011) Effects of hydroxypropyl-beta-cyclodextrin on cell growth, activity, and integrity of steroid-transforming Arthrobacter simplex and Mycobacterium sp. Appl Microbiol Biotechnol 90:1995–2003. https://doi.org/10.1007/s00253-011-3214-6
Singer Y, Shity H, Bar R (1991) Microbial transformations in a cyclodextrin medium. Part 2. Reduction of androstenedione to testosterone by Saccharomyces cerevisiae. Appl Microbiol Biotechnol 35:731–737
Singh M, Sharma R, Banerjee U (2002) Biotechnological applications of cyclodextrins. Biotechnol Adv 20:341–359
Stammen S, Müller BK, Korneli C, Biedendieck R, Gamer M, Franco-Lara E, Jahn D (2010) High-yield intra-and extracellular protein production using Bacillus megaterium. Appl Environ Microbiol 76:4037–4046
Stancu MM (2018) Isolation and characterization of Bacillus megaterium IBBPo17 a solvent-tolerant bacterium. Waste Biomass Valoriz. https://doi.org/10.1007/s12649-018-0354-2
Straathof AJJ, Panke S, Schmid A (2002) The production of fine chemicals by biotransformations. Curr Opin Biotechnol 13:548–556
Torres S, Pandey A, Castro GR (2011) Organic solvent adaptation of Gram positive bacteria: applications and biotechnological potentials. Biotechnol Adv 29:442–452. https://doi.org/10.1016/j.biotechadv.2011.04.002
Valisena S, Palumbo M, Parolin C, Palu G, Meloni GA (1990) Relevance of ionic effects on norfloxacin uptake by Escherichia coli. Biochem Pharmacol 40:431–436. https://doi.org/10.1016/0006-2952(90)90540-2
Vary PS, Biedendieck R, Fuerch T, Meinhardt F, Rohde M, Deckwer WD, Jahn D (2007) Bacillus megaterium–from simple soil bacterium to industrial protein production host. Appl Microbiol Biotechnol 76:957–967. https://doi.org/10.1007/s00253-007-1089-3
Vermue M, Sikkema J, Verheul A, Bakker R, Tramper J (1993) Toxicity of homologous series of organic solvents for the gram-positive bacteria Arthrobacter and Nocardia sp. and the gram-negative bacteria Acinetobacter and Pseudomonas sp. Biotechnol Bioeng 42:747–758. https://doi.org/10.1002/bit.260420610
Walton AZ, Stewart JD (2002) An efficient enzymatic Baeyer-Villiger oxidation by engineered Escherichia coli cells under non-growing conditions. Biotechnol Prog 18:262–268
Walton AZ, Stewart JD (2004) Understanding and improving NADPH-dependent reactions by nongrowing Escherichia coli cells. Biotechnol Prog 20:403–411
Wang M, Zhang L, Shen Y, Ma Y, Zheng Y, Luo J (2009) Effects of hydroxypropyl-β-cyclodextrin on steroids 1-en-dehydrogenation biotransformation by Arthrobacter simplex TCCC 11037. J Mol Catal B Enzyme 59:58–63. https://doi.org/10.1016/j.molcatb.2008.12.017
Westfall PJ, Pitera DJ, Lenihan JR, Eng D, Woolard FX, Regentin R, Horning T, Tsuruta H, Melis DJ, Owens A, Fickes S, Diola D, Benjamin KR, Keasling JD, Leavell MD, McPhee DJ, Renninger NS, Newman JD, Paddon CJ (2012) Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc Natl Acad Sci USA 109:E111–E118. https://doi.org/10.1073/pnas.1110740109
Williams KJ, Piddock LJ (1998) Accumulation of rifampicin by Escherichia coli and Staphylococcus aureus. J Antimicrob Chemother 42:597–603. https://doi.org/10.1093/jac/42.5.597
Xu S, Dai WG (2013) Drug precipitation inhibitors in supersaturable formulations. Int J Pharm 453:36–43. https://doi.org/10.1016/j.ijpharm.2013.05.013
Yonekura L, Sun H, Soukoulis C, Fisk I (2014) Microencapsulation of Lactobacillus acidophilus NCIMB 701748 in matrices containing soluble fibre by spray drying: technological characterization, storage stability and survival after in vitro digestion. J Funct Foods 6:205–214. https://doi.org/10.1016/j.jff.2013.10.008
Yong C, Devine SM, Gao X, Yan A, Callaghan R, Capuano B, Scammells PJ (2019) A novel class of N-sulfonyl and N-sulfamoyl noscapine derivatives that promote mitotic arrest in cancer cells. ChemMedChem 14:1968–1981
Zangar RC, Davydov DR, Verma S (2004) Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol Appl Pharmacol 199:316–331. https://doi.org/10.1016/j.taap.2004.01.018
Zehentgruber D, Dragan CA, Bureik M, Lutz S (2010) Challenges of steroid biotransformation with human cytochrome P450 monooxygenase CYP21 using resting cells of recombinant Schizosaccharomyces pombe. J Biotechnol 146:179–185. https://doi.org/10.1016/j.jbiotec.2010.01.019
Zhang HM, Li Z, Uematsu K, Kobayashi T, Horikoshi K (2008) Antibacterial activity of cyclodextrins against Bacillus strains. Arch Microbiol 190:605–609. https://doi.org/10.1007/s00203-008-0415-1
Acknowledgements
The authors would like to thank the Mass Spectrometry and Proteomics Facility at the Bio21 Institute (Melbourne) for technical assistance and access to analytical equipment. This work was supported by Sun Pharmaceutical Industries Ltd and The University of Melbourne. LR was supported by an Australian Government Research Training Program Scholarship.
Author information
Authors and Affiliations
Contributions
LR performed the experiments. TB, AJ, GS and SG helped with experimental design and provided technical advice. LR and SG wrote the manuscript with input from all authors.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Richards, L., Jarrold, A., Bowser, T. et al. Cytochrome P450-mediated N-demethylation of noscapine by whole-cell biotransformation: process limitations and strategies for optimisation. J Ind Microbiol Biotechnol 47, 449–464 (2020). https://doi.org/10.1007/s10295-020-02283-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-020-02283-7