
Abstract
Butanol production from agricultural residues is the most promising alternative for fossil fuels. To reach the economic viability of biobutanol production, both glucose and xylose should be utilized and converted into butanol. Here, we engineered a dual-operon-based synthetic pathway in the genome of E. coli MG1655 to produce n-butanol using CRISPR/Cas9 technology. Further deletion of competing pathway followed by fed-batch cultivation of the engineered strain in a bioreactor with glucose-containing complex medium yielded 5.4 g/L n-butanol along with pyruvate as major co-product, indicating a redox imbalance. To ferment xylose into butanol in redox-balanced manner, we selected SSK42, an ethanologenic E. coli strain engineered and evolved in our laboratory to produce ethanol from xylose, for integrating synthetic butanol cassette in its genome via CRISPR/Cas9 after deleting the gene responsible for endogenous ethanol production. The engineered plasmid- and marker-free strain, ASA02, produced 4.32 g/L butanol in fed-batch fermentation in completely defined AM1–xylose medium.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biofuels produced from lignocellulosic biomass has been considered as a feasible alternative to fossil fuels. The exhaustion of fossil fuels combined with excessive greenhouse gas emissions during its combustion have attracted much attention to produce required energy from biomass [22]. As a clean and renewable fuel source, there was a growing interest in the production of bioethanol in recent years and it has been widely recognized as a typical biofuel; however, ethanol is not considered as an ideal substitute for gasoline. Biobutanol is a renewable alternative that possesses various important advantages compared to ethanol, including higher energy content, hydrophobicity and octane number, and lower vapor pressure, which provide better blending ability with gasoline at any fraction. In addition, it can be used directly in conventional engines without any modification [8, 26, 35].
Biobutanol is efficiently produced from renewable resources (biomass) via ABE fermentation process using Clostridium acetobutylicum [33]. Due to limited genetic manipulation system and complex physiology of Clostridium, other bacterial strains, such as E. coli, have been developed to produce biobutanol. E. coli, a non-native user-friendly host was first engineered with a synthetic butanol production pathway from Clostridium acetobutylicum including seven genes thl, hbd, crt, bcd, etfA, etfB, and adhE2, to produce one molecule of butanol from two molecules of acetyl-CoA in six steps. However, the engineered strain produced less than 1 g/L butanol [1]. Recently, various works was reported on the hetero-production of butanol in different E. coli strains with much progress in strain improvement [7, 12, 27].
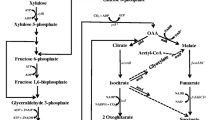
Lignocellulosic biomass derived from agricultural residues is the most abundant renewable source of sugars that does not compete with foodstuffs, and thus it is an excellent feedstock for biobutanol production [32]. Cellulose (35–50%) and hemicellulose (25–30%) are the major components of lignocellulosic biomass, and whereas glucose is the main monosaccharide obtained by hydrolysis of the cellulosic fraction, xylose is the second most abundant monosaccharide resulting from the hydrolysis of hemicellulose [31]. Therefore, for the economic viability of bulk molecules such as biobutanol, it is important that both these sugars of lignocellulosic biomass are utilized during their production. However, the microorganisms commonly used in industry do not have the ability to ferment and convert xylose into useful products, and only few reports are available on the investigation of fermentation for butanol production using xylose as the sole carbon source [35]. Therefore, more efforts are still needed in terms of expanding substrate range, utilizing low-cost defined medium, avoiding the use of plasmid-based expression and improving yield, productivity and titer of butanol. The question that we tried to address in our current study is that can we engineer an ethanologenic strain of E. coli, which had been previously engineered and evolved to efficiently convert xylose in defined medium into ethanol without the use of any foreign gene, into a butanol producer. Many reports previously used plasmid-based expression system to obtain much higher butanol production [1, 11, 13, 34]. However, we believe bulk molecules such as butanol should not be produced using plasmid-based system that has an inherent issue of instability upon scale-up along with the use of expensive antibiotics. We thus used CRISPR/Cas9 tool to integrate a large portion of DNA fragment carrying butanol pathway into genome. However, even when we use the similar pathway characterized in some other strains/organisms, assembling them in the new strains/organisms always poses new challenges. Thus, to prove the hypothesis in terms of the expression of enzymes and level of butanol production, the pathway integration was first performed in a well-studied system, i.e., E. coli MG1655. After achieving the success in this strain, we engineered the genome of an efficient xylose-utilizing host, i.e., E. coli SSK42 (Fig. 1), and shown for the first time the production of butanol from xylose as a sole carbon source in defined medium.

Schematic representation of butanol production pathway in engineered E. coli ASA02 strain. The butanol production pathway from acetyl-CoA consists of six enzymatic steps as highlighted in dark yellow. THL acetyl transferase, HBD β-hydroxybutyryl-CoA dehydrogenase, CRT 3-hydroxybutyryl-CoA dehydratase, TER trans-enoyl-coenzyme A (CoA) reductase, BAD/BDH butyraldehyde/butanol dehydrogenase, PYK pyruvate kinase, PgapAPDH pyruvate dehydrogenase operon expressed under the promoter of glyceraldehyde-3-phosphate dehydrogenase gene (present in background E. coli SSK42 host)
Materials and methods
Bacterial strains and plasmids
Escherichia coli strains and plasmids used in this study are listed in Table 1.
Plasmid construction for butanol production via acetoacetyl-CoA reduction pathway
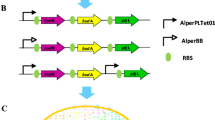
pET–BuOH construct was synthesized commercially (GenScript, USA), which included codon-optimized n-butanol pathway genes, i.e., hbd [coding for beta-hydroxybutyryl-CoA dehydrogenase (HBD)], crt [coding for 3-hydroxybutyryl-CoA dehydratase (CRT)] and adhE2 [coding for butyraldehyde/butanol dehydrogenase (BAD/BDH)] from Clostridium acetobutylicum, ter [coding for trans-enoyl-coenzyme A (CoA) reductase (TER)] from Treponema denticola and atoB [coding for acetyl-CoA acetyltransferase (THL)] from E. coli. The whole pathway genes were distributed in two operons, both operons being under the control of two separate T5 promoters. First operon contained atoB, hbd and crt, while the second operon included adhE2 and ter genes. We also included 600-bp homology arms of an intergenic region SS9 (safe site 9) of E. coli genome, which was previously found to provide high integration efficiency as well as an expression level of heterologous gene [2], to the 5′ and 3′ end of the construct to facilitate genomic integration via CRISPR/Cas9 technology. The whole synthetic construct containing two operons along with homology arms was cloned at BamHI/HindIII in pET28a( + ) vector as shown in Fig. 2.

The optimized synthetic construct of essential set of genes for butanol production. The construct contains two operons under control of T5 promoter. The first operon contains atoB, hbd and crt genes, and the second operon contains adhE2 and ter genes. A homology region of 600 bp on both sides of the construct was introduced to facilitate integration at intergenic region SS9 of the genome. The synthetic construct was cloned and expressed using the backbone of vector pET28a(+)
Genome engineering via CRISPR/Cas9 in E. coli MG1655 and SSK42 strains
Single colony of freshly transformed E. coli MG1655 or SSK42 cells with pCas plasmid (Catalog no. 62225, Addgene) was inoculated in LB and induced with 10 mM arabinose for λ red recombinase gene induction before processing for making electro-competent cells. 50 µL of the electro-competent cells were mixed with 100 ng of SS9_RNA plasmid (Catalog no. 71656, Addgene) and 400 ng of linear donor DNA (BamHI/HindIII digested product of pET–BuOH) and electroporation was performed in 1-mm Gene Pulser cuvette (Bio-Rad) at 2.0 kV. After the electric pulse, cells were suspended immediately in 1 mL of LB medium and incubated at 30 °C for 1 h before spreading onto LB agar plate containing kanamycin (50 mg/L) and ampicillin (100 mg/L). The plates were further incubated overnight at 30 °C and positive transformants were identified by colony PCR.
For curing SS9_RNA plasmid, the genome-edited colonies harboring both the plasmids were inoculated in 2 mL of LB medium containing kanamycin (50 mg/L) and incubated for 8–16 h at 37 °C, diluted, and spread onto LB agar plates containing kanamycin (50 mg/L). The colonies were confirmed as cured by determining their sensitivity to ampicillin (100 mg/L). pCas plasmid was cured by growing the colonies overnight at 37 °C non-selectively and screening for loss in resistance towards kanamycin.
Gene knockout
Single-gene knockout Keio collection strains obtained from the Coli Genetic Stock Center (CGSC), Yale University, USA, were used for gene deletion in this study. Knockout of adhE gene was generated by phage (P1) transduction method [17] using allelic replacement to insert 1.9-kb FRT-flanked kanamycin cassette, which was confirmed by PCR. This cassette was excised with the help of flippase using pCP20 plasmid.
Growth and fermentation experiments
For E. coli MG1655-based modified strains, small volume cultivation for n-butanol production was performed by inoculating a single colony from freshly streak LB plate to 5 mL LB medium with appropriate antibiotics in culture tube and grown for overnight at 37 °C and 180 rpm. 1% of this primary culture was used to inoculate 10 mL Terrific Broth (TB) medium supplemented with 1% glucose and 0.1 mM IPTG in 15-mL falcon tube, followed by incubation at 37 °C on a Gyrorotator. Cultures were analyzed for n-butanol production after 48 h of cultivation. For fed-batch fermentation experiment, KJK01 strain was used for n-butanol production in a 5-L stirred bioreactor (Applikon Biotechnology). We followed the strategy published by Shen et al. [25] for butanol production where the authors showed maximum butanol production in Terrific Broth (TB)-based medium. Initially, we grew a single colony from freshly streak LB plate in 5 mL LB medium at 37 °C and 180 rpm for overnight. Secondary culture was prepared by inoculating 1% of primary culture to 300 mL of TB medium supplemented with 1% glucose and grown at 37 °C until OD600 reached to 2–3. The grown secondary culture was used as seed culture to inoculate the bioreactor with working volume of 3 L, having TB media along with 20 g/L glucose and 0.1 mM IPTG. The cells were first cultivated at 37 °C under aerobic conditions in batch mode until the OD600 reached ~ 5 in 6 h. Then, 2 VVM (volume of gas per volume of liquid per minute) of nitrogen was bubbled through the bioreactor to switch to anaerobic conditions. Upon the anaerobic switch, intermittent linear feeding of glucose solution (500 g/L) was initiated to maintain a glucose concentration between 5 and 10 g/L. Samples were collected at different time interval for measuring cell density and metabolite concentration.
For the ASA02 strain where butanol pathway had been integrated into the genome of E. coli SSK42, the small-scale test-tube experiments were conducted in Hungate tubes (18 mL capacity). A pool of colonies growing on AM1 agar plate containing 2% xylose was used to inoculate 12 mL AM1 liquid medium in Hungate tube and grown for 4–5 h at 37 °C for primary inoculum preparation. The grown cells from here were used to inoculate fresh 12 mL AM1 liquid medium containing different concentrations of xylose and IPTG as mentioned in results section with an initial OD600 of 0.1. The cultures were grown at 37 °C and samples were collected at various time intervals for measuring cell growth and metabolites. Batch and fed-batch cultivations were performed using ASA02 strain in a 500-mL bioreactor (Applikon Biotechnology). For inoculum preparation, a pool of colonies was suspended in Hungate tubes in duplicates, each containing 12 mL of AM1 medium with 2% xylose, and incubated at 37 °C in a Gyrorotator for 6 h. For the inoculation in the bioreactor, the inoculum was centrifuged at 5000 rpm for 5 min and cell pellet was resuspended in 1 mL of AM1 medium and inoculated in the bioreactor containing 300 mL of AM1 medium containing different concentrations of xylose with an initial OD600 of ~ 0.1. The bioreactor was operated under an agitation speed of 500 rpm at 37 °C. Air was supplied into the headspace to maintain microaerobic condition. 2 N KOH was supplemented to adjust pH at 6.8. Samples were collected from the bioreactor for cell growth, sugar and metabolite analyses. All the experiments were repeated at least twice and representative fermentation profiles were plotted.
Analytical techniques
Cell density was determined by measuring absorbance at 600 nm (OD600) using a spectrophotometer (GE Healthcare). Xylose, butanol and other fermentation products were analyzed by a high-performance liquid chromatography system (HPLC) equipped with an Aminex HPX 87H (300 × 7.8 mm) column and RI detector (Agilent technologies) using the method previously described [15]. Samples were collected from the culture and cleared from biomass by centrifugation at 13,400 rpm for 1 min. The supernatant was then diluted with 4 mM H2SO4, filtered via 0.22-µm syringe filter, and 10 μL of filtered sample was used for injection via the autosampler. The column temperature was maintained at 40 °C and 4 mM H2SO4 was used as mobile phase at flow rate of 0.3 mL/min. Reference standards (Absolute Standards, USA) of 1 g/L were used to estimate the concentration of sugar and fermentation products in each sample.
Results
Expression of optimized pathway for butanol production in E. coli
We considered Clostridial-based fermentative pathway for n-butanol production in E. coli, with the exception of first pathway enzyme acetyl-CoA acetyl transferase (THL) from E. coli itself and the intermediate enzyme trans-enoyl-coenzyme A (CoA) reductase (TER) from the T. denticola (Fig. 1). These alternative enzymes from non-Clostridial sources were shown earlier to be more efficient in n-butanol production under fermentative condition [1, 3]. All five pathway genes were codon optimized for an optimal expression in E. coli and were segregated under two operons, first operon contained atoB, hbd and crt genes and the second operon contained adhE2 and ter genes (Fig. 2). Both operons were under the control of the IPTG-inducible T5 promoter. A homology arm of 600 bp of an intergenic region SS9 (safe site 9) was introduced to the 5′ and 3′ ends of the construct for genomic integration via CRISPR/Cas9 technique. The whole synthetic construct was cloned and expressed using the backbone of vector pET28a(+) (Fig. 2).
To check the efficacy of the optimized pathway for butanol production, we used pET–BuOH construct to transform E. coli MG1655 and cultivated in TB medium supplemented with 1% glucose at 37 °C. We observed 64 mg/L of butanol titer in the medium after 48 h of cultivation (Fig. 3), which confirmed the functionality of the pathway enzymes encoded by the synthetic construct. However, we also observed the formation of high level of byproducts such as acetate, lactate, succinate and ethanol in the medium (Additional file 1: Table S1). To minimize these byproducts, we considered E. coli LA07 strain, which had deletion of pta, adhE, frdA and ldhA genes encoding phosphate acetyltransferase, alcohol dehydrogenase, fumarate reductase and lactate dehydrogenase to prevent the formation of acetate, ethanol, succinate and lactate, respectively, in MG1655 [12]. When E. coli LA07 was transformed with pET–BuOH plasmid, we observed 1.34 g/L butanol, a value that was 21-fold higher than that obtained using parent MG1655 strain.

Comparison of butanol productions in engineered E. coli via acetoacetyl-CoA reduction pathway. Cells were grown in Terrific Broth medium supplemented with 1% glucose at 37 °C in test tubes for 48 h
Genomic integration of butanol pathway via CIRSPR/Cas9
Genomic integration of heterologous constructs provides long-term stability and reduces cell to cell variability in copy number and expression levels, making such strains more suitable for industrial-scale processes or downstream engineering compared to plasmid-based system. We, therefore, integrated the optimized pathway cassette in the genome of LA07 strain with the help of CRISPR/Cas9 technique. We aimed for markerless integration to avoid concern related to transfer of resistance marker to the non-target species in case of scape of engineered strain during large-scale production. The optimized cassette was synthesized along with 600-bp homology region on both sides to facilitate integration at intergenic region SS9 of the genome. This cassette was supplied to the cells as linear BamH1/HindIII-digested DNA fragment via electroporation (Fig. 2). sgRNA specific to SS9 site was provided via plasmid SS9_RNA while Cas9 for a double-strand break in the SS9 region and λ red recombinase for integrating the optimized cassette at the cleavage site were provided via pCas plasmid. The integration of the n-butanol cassette in the genome of LA07 was confirmed through PCR and both the plasmids were removed as mentioned in “Materials and methods” to make the engineered strain completely marker free.
The final strain KJK01, harboring butanol pathway in the genome, was cultivated in TB medium supplemented with 1% glucose at 37 °C and metabolite formation was analyzed after 48 h of cultivation. To our surprise, we observed very similar titer (1.1 g/L) of butanol in the single-copy genome-integrated butanol pathway compared to the titer (1.34 g/L) obtained via multi-copy plasmid, suggesting randomly increasing the copy of number of pathway genes is not always beneficial for the metabolite production (Fig. 3).
Assessment of genomic integrated butanol pathway in bioreactor
The genome-integrated butanol pathway strain E. coli KJK01 was further used to produce butanol in a 5-L bioreactor. We first grew the cells under aerobic conditions in batch mode until the OD600 reached about 5 in 6 h. The cultivation condition was then switched to an anaerobic state. We observed a slow growth of cells after anaerobic switch of the culture (Fig. 4), but butanol production increased during the anaerobic phase from 0.76 g/L at 6 h to 5.4 g/L at 27 h. Moreover, we observed significant glucose consumption during this period, which seemed to be majorly diverted towards pyruvate production with the titer of 17.5 g/L. This was not surprising because under anaerobic condition the pathway to butanol from glucose consumes four NADH while only two NADH is produced. To balance this loss of NADH, the cell produces more of pyruvate that requires no NADH for its production. We also observed an equal quantity of ethanol (5.4 g/L) even when the engineered strain had the deletion of endogenous alcohol dehydrogenase gene (adhE). This is due to the fact that Clostridial butyraldehyde/butanol dehydrogenase can also recognize acetyl-CoA as substrate in addition to butyryl-CoA and produce ethanol in addition to butanol. Besides, we also observed some butyrate production (1.8 g/L). The butanol was produced at maximum productivity of 0.39 g/L/h (5.23 mmol/L/h) and product yield of 0.13 g/g glucose, which was 31% of theoretical maximum (Table 2).

Fed-batch fermentation profile of E. coli KJK01 strain. Bacterial strain was cultivated in TB medium with glucose in a bioreactor under aerobic condition until the OD600 reached ~ 5 in 6 h; then the cultivation was switched to anaerobic condition
Engineering E. coli strain for butanol production from xylose
For producing butanol from xylose, we considered a xylose-fermenting ethanologenic E. coli strain SSK42 as a platform host for metabolic engineering. The SSK42 was earlier constructed in our laboratory to produce ethanol from glucose and xylose by engineering the promoter of pyruvate dehydrogenase operon and deleting the genes responsible for side products [20]. This strain was further evolved on C5 and C6 sugars to improve the rate of substrate consumption and product formation in the defined medium [16]. The evolved ethanologenic strain SSK42 has a balanced redox pathway for the ethanol production from glucose and xylose. Since electron requirement for both ethanol and butanol production is same, we decided to engineer SSK42 strain to produce butanol from xylose as a sole carbon source. We first deleted endogenous adhE gene of SSK42 via P1 phage transduction and then integrated synthetic butanol pathway cassette in its genome using CRISPR/Cas9 technique to divert carbon flux towards butanol (Fig. 1). The new engineered strain ASA02 was further evaluated for its ability to produce butanol. Induction of butanol operons using 0.01 mM IPTG was found to be more appropriate for butanol production (Fig. 5a). Upon growing the engineered cells at various xylose concentrations in the test tube, 1% xylose concentration was found to be optimal for butanol production (Fig. 5b). We did not observe significant pyruvate production by the ASA02 strain but we did notice higher ethanol production.

Comparison of the effect of different IPTG and xylose concentrations on the production of alcohol in E. coli ASA02 strain. a Bacterial cells were grown in Hungate tubes in the presence of 0.01 mM and 0.1 mM IPTG at 37 °C for 48 h. b Bacterial cells were grown in Hungate tubes with 1%, 3% and 5% xylose at 37 °C for 48 h
Xylose fermentation to butanol at bioreactor level
Different fermentation strategies were performed to produce butanol from xylose sugar in defined AM1 medium. Based on the experience from the parent strain of ASA02, i.e., SSK42, for ethanol production [16], we first grew the engineered strain in AM1 + 10% xylose in the bioreactor under microaerobic condition. Cells continued to grow until 72 h and reached stationary phase at ~ OD600 of 2.8 (Fig. 6a). Soon after, butanol also reached its maxima of 2.1 g/L at 84 h of cultivation. However, xylose consumption, ethanol formation and acetic acid formation continued beyond 84 h.

Batch fermentation profile of E. coli ASA02 strain. a Bacterial cells were grown in AM1 medium with xylose under microaerobic condition in a bioreactor. b Bacterial cells were grown in AM1 medium with xylose in a bioreactor under aerobic condition for 19 h then shifted to anaerobic condition
Since it appeared from the above experiment that butanol production is growth associated, we attempted to enhance the growth of the engineered strain by initially growing under aerobic condition and then switching to anaerobic condition for metabolite production. As expected, the cells grew faster and reached to OD600 of 8.06 in 19 h, with no further increase upon switching to anaerobic condition (Fig. 6b). However, higher growth via two-stage fermentation did not lead to higher butanol production, which only reached to 1.53 g/L titer after 55 h of fermentation.
Taking clue from test tube experiment about low initial xylose concentration (1%) being beneficial for butanol production (Fig. 5b), we performed fed-batch fermentation where we initiated batch fermentation in 1% xylose and then further added xylose intermittently to maintain the level ~ 0.5–1% in the bioreactor. We first tested the impact of airflow rate on butanol production under microaerobic condition. This experiment was conducted by supplying air flow in the headspace at the rate ranging from 0.03 to 1.66 VVM (Additional file 1: Figure S1). The butanol production increased when the airflow rate was increased from 0.03 to 0.08 VVM and then decreased upon a subsequent increase in the airflow rate. We observed a consistent increase in butanol formation throughout the fermentation at 0.08 VVM of airflow rate, which reached to 4.3 g/L at 188 h with 0.13 g/g xylose yield (Fig. 7). The curve of xylose consumption and butanol formation almost overlapped each other, indicating similar product yield throughout the fermentation. Under this condition, ethanol concentration did not go higher than butanol concentration, as was observed in the previous two types of fermentation in AM1 defined medium.

Fed-batch fermentation profile of E. coli ASA02 strain. Bacterial cells were grown in AM1 medium with xylose under microaerobic condition in a bioreactor
Discussion
Engineering of a biosynthetic pathway in a non-native producer not only needs the optimal expression of the pathway genes but also require the deletion of key enzymes in the competing pathways. The first committed enzyme of any biosynthetic pathway is important to drive the carbon flux towards that pathway. In the fermentative pathway of n-butanol production, the first committed reaction is condensation of two acetyl-CoA molecules. For this reaction, acetyl-CoA acetyltransferase (THL) of E. coli encoded by atoB showed much higher specific enzyme activity (1078 U/mg vs 216 U/mg) and the butanol titer (threefold higher) compared to the Clostridial THL [1]. For the other reactions in the pathway, β-hydroxybutyryl-CoA dehydrogenase (HBD), 3-hydroxybutyryl-CoA dehydratase (CRT) and butyraldehyde/butanol dehydrogenase (BAD/BDH) from C. acetobutylicum have been reported to be best for their functions [4, 10, 30]. However, for converting crotonyl-CoA to butyryl-CoA, replacement of butyryl-CoA dehydrogenase enzyme of C. acetobutylicum, which catalyzes this as a reversible reaction and uses ferredoxin as electron donor, with trans-enoyl-coenzyme A (CoA) reductase (TER) from T. denticola, which catalyzes the same reaction but irreversibly and uses NADH as electron donor, resulted in significantly high butanol titer [5].
We, therefore, in our effort to make n-butanol from xylose as a carbon source, we considered THL from E. coli, HBD, CRT and BAD/BDH from C. acetobutylicum and TER from T. denticola. Gene encoding these enzymes were codon optimized and assembled under two operons for optimal expression. This was done since it has been reported previously that the expression level decreases with increase in the number of CDS in an operon, with first CDS being expressed most [29]. Within the two operons, we placed atoB first in one operon since its product catalyzes the first committed step in the butanol pathway and adhE2 first in the second operon since its product catalyzes last reaction in the pathway, and their higher expression is likely to drive the carbon flux more towards this pathway.
As well known about functionality of metabolic pathway of E. coli during fermentation [20], the butanol titer was outcompeted by the titers of various metabolites, such as ethanol, acetate, lactate and succinate. Therefore, the use of engineered strain LA07 having deletion in the genes of the pathways responsible for these fermentative products led to an improvement in butanol titer by 21-fold. Further, we believe bulk molecules such as butanol should not be produced using plasmid-based system as it has an inherent issue of instability upon scale-up as well as the requirement for use of expensive antibiotics. We thus used CRISPR/Cas9 tool to integrate a large portion of DNA fragment carrying butanol pathway into genome of LY07. This genome engineering effort yielded similar butanol titer as was achieved via plasmid construct. This observation was highly encouraging considering that the complete pathway was integrated in the genome only as a single copy while it is present as multiple copies inside the cell when expressed via plasmid pET28a. Our results also confirmed the earlier finding that the integration of heterologous gene cassette in the intergenic region, SS9, was suitable for good expression levels [2]. But we did observe high quantity of pyruvate accumulation due to imbalanced reducing equivalents, indicating the need to construct a strain that has redox-balanced pathway for butanol production.
To achieve the goal of producing butanol from xylose via a redox-balanced pathway, we considered xylose-fermenting E. coli strain, SSK42, that has the ability to produce a high concentration of ethanol [16]. The advantage of this strain is that it has no foreign gene in its genome and it was engineered in our laboratory to produce ethanol from glucose and xylose by modulating the expression of pyruvate dehydrogenase under anaerobic condition and deleting the fermentative pathway genes [16]. In this way, a redox-balanced pathway was achieved for producing ethanol from either glucose or xylose. We realized that the requirement of number of reducing equivalence for producing one molecule butanol from xylose is same as that of producing two molecules of ethanol (Fig. 1); thus, it should be possible to use SSK42 as a host for butanol production in a redox-balanced manner. We, therefore, replaced ethanol-producing pathway with butanol-producing pathway in the genome of E. coli SSK42 by deleting adhE gene and integrating synthetic butanol pathway construct. The NADH-dependent AdhE enzyme of E. coli is a bifunctional enzyme composed of an aldehyde dehydrogenase and an alcohol dehydrogenase that can catalyze the reduction of acetyl-CoA to acetaldehyde and acetaldehyde to ethanol, and this enzyme is a major catalyst for ethanol formation in E. coli [12]. Deleting gene for this enzyme should abrogate the ability of E. coli to produce ethanol. A genome-engineered ASA02 strain with the synthetic butanol pathway genes produced ~ 1 g/L of butanol from xylose sugar in minimal AM1 medium (Fig. 5). We did not observe significant production of pathway intermediate pyruvate in this strain, indicating balancing of redox equivalence. However, even after deleting adhE gene, we detected relatively high amount of ethanol (up to 2.2 g/L) in SSK42 ΔadhE. This could be because the product of adhE2, i.e., C. acetobutylicum butyraldehyde/butanol dehydrogenase, can also recognize acetyl-CoA as substrate in addition to butyryl-CoA and produce ethanol [24]. Another important concern for using xylose as a sole carbon source under anaerobic condition is low ATP yield (0.67 mol ATP/mole xylose) [28], leading to low cell growth. One way to overcome this is by growing the cells under microaerobic condition [20]. However, it was important to find out appropriate flow rate of air that would not compromise the yield of the reduced product, such as butanol. In the range of airflow rate tested between 0.03 and 1.66 VVM, 0.08 VVM was optimal for butanol production (Additional file 1: Figure S1). The butanol titer reached up to 4.32 g/L in the fed-batch fermentation, and along with ethanol, 8.5 g/L of total solvent was produced with 58% of theoretical yield (Table 2). The work reported here is the first study where the strain engineering effort led to significant production of butanol from xylose in minimal media.
Recently, a synthetic consortium of two E. coli strains was developed for co-utilization of glucose and xylose to produce n-butanol [24], but pure cultures are more desirable during industrial fermentations. Therefore, further engineering for co-utilization of different sugars typically present in lignocellulosic biomass, such as glucose, xylose, and arabinose, will be beneficial. Previous work have also demonstrated a significant improvement in the product titer upon laboratory evolution [6]. Adaptive laboratory evolution (ALE), where a strain is selected with desired phenotype from randomly generated variants, has been applied to improve tolerance and performance of E. coli for a product that is difficult to achieve via rational metabolic engineering [18, 19, 23]. Similarly, toxicity of the product in the culture media has been reduced by performing extractive fermentation via layering culture media with dodecane [9, 27] or in situ removal of the product [14]. These are the future courses of actions that are likely to improve the butanol titer by the engineered ASA02 strain to industrially acceptable level.
Conclusions
In our effort to make industry friendly process for n-butanol production, we first engineered a plasmid- and marker-free E. coli strain with the help of CRISPR/Cas9 technique to produce 5.4 g/L of n-butanol in complex medium with glucose as a carbon source. The whole engineering effort was then replicated into an ethanologenic strain to produce butanol via redox-balanced pathway in minimal medium with xylose as a sole carbon source.
References
Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, Chou KJ, Hanai T, Liao JC (2008) Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng 10:305–311. https://doi.org/10.1016/j.ymben.2007.08.003
Bassalo MC, Garst AD, Halweg-Edwards AL, Grau WC, Domaille DW, Mutalik VK, Arkin AP, Gill RT (2016) Rapid and efficient one-step metabolic pathway integration in E. coli. ACS Synth Biol 5:561–568. https://doi.org/10.1021/acssynbio.5b00187
Bond-Watts BB, Bellerose RJ, Chang MC (2011) Enzyme mechanism as a kinetic control element for designing synthetic biofuel pathways. Nat Chem Biol 7:222–227. https://doi.org/10.1038/nchembio.537
Boynton ZL, Bennet GN, Rudolph FB (1996) Cloning, sequencing, and expression of clustered genes encoding beta-hydroxybutyryl-coenzyme A (CoA) dehydrogenase, crotonase, and butyryl-CoA dehydrogenase from Clostridium acetobutylicum ATCC 824. J Bacteriol 178:3015–3024
Bond-Watts BB, Bellerose RJ, Chang MC (2011) Enzyme mechanism as a kinetic control element for designing synthetic biofuel pathways. Nat Chem Biol 7:6
Choi KR, Jang WD, Yang D, Cho JS, Park D, Lee SY (2019) Systems metabolic engineering strategies: integrating systems and synthetic biology with metabolic engineering. Trends Biotechnol. https://doi.org/10.1016/j.tibtech.2019.01.00310.1016/j.tibtech.2019.01.003
Dellomonaco C, Clomburg JM, Miller EN, Gonzalez R (2011) Engineered reversal of the beta-oxidation cycle for the synthesis of fuels and chemicals. Nature 476:355–359. https://doi.org/10.1038/nature10333
Durre P (2007) Biobutanol: an attractive biofuel. Biotechnol J 2:1525–1534. https://doi.org/10.1002/biot.200700168
Fatma Z, Hartman H, Poolman MG, Fell DA, Srivastava S, Shakeel T, Yazdani SS (2018) Model-assisted metabolic engineering of Escherichia coli for long chain alkane and alcohol production. Metab Eng 46:1–12. https://doi.org/10.1016/j.ymben.2018.01.002
Fontaine L, Meynial-Salles I, Girbal L, Yang X, Croux C, Soucaille P (2002) Molecular characterization and transcriptional analysis of adhE2, the gene encoding the NADH-dependent aldehyde/alcohol dehydrogenase responsible for butanol production in alcohologenic cultures of Clostridium acetobutylicum ATCC 824. J Bacteriol 184:821–830
Garza E, Zhao J, Wang Y, Wang J, Iverson A, Manow R, Finan C, Zhou S (2012) Engineering a homobutanol fermentation pathway in Escherichia coli EG03. J Ind Microbiol Biotechnol 39:1101–1107. https://doi.org/10.1007/s10295-012-1151-8
Han GH, Seong W, Fu Y, Yoon PK, Kim SK, Yeom SJ, Lee DH, Lee SG (2017) Leucine zipper-mediated targeting of multi-enzyme cascade reactions to inclusion bodies in Escherichia coli for enhanced production of 1-butanol. Metab Eng 40:41–49. https://doi.org/10.1016/j.ymben.2016.12.012
Inui M, Suda M, Kimura S, Yasuda K, Suzuki H, Toda H, Yamamoto S, Okino S, Suzuki N, Yukawa H (2008) Expression of Clostridium acetobutylicum butanol synthetic genes in Escherichia coli. Appl Microbiol Biotechnol 77:1305–1316. https://doi.org/10.1007/s00253-007-1257-5
Jang YS, Malaviya A, Lee SY (2013) Acetone-butanol-ethanol production with high productivity using Clostridium acetobutylicum BKM19. Biotechnol Bioeng 110:1646–1653. https://doi.org/10.1002/bit.24843
Jawed K, Mattam AJ, Fatma Z, Wajid S, Abdin MZ, Yazdani SS (2016) engineered production of short chain fatty acid in escherichia coli using fatty acid synthesis pathway. PLoS One 11:e0160035. https://doi.org/10.1371/journal.pone.0160035
Jilani SB, Venigalla SSK, Mattam AJ, Dev C, Yazdani SS (2017) Improvement in ethanol productivity of engineered E. coli strain SSY13 in defined medium via adaptive evolution. J Ind Microbiol Biotechnol 44:1375–1384. https://doi.org/10.1007/s10295-017-1966-4
Mattam AJ, Yazdani SS (2013) Engineering E. coli strain for conversion of short chain fatty acids to bioalcohols. Biotechnol Biofuels 6:128. https://doi.org/10.1186/1754-6834-6-128
Mukhopadhyay A (2015) Tolerance engineering in bacteria for the production of advanced biofuels and chemicals. Trends Microbiol 23:498–508. https://doi.org/10.1016/j.tim.2015.04.008
Mundhada H, Seoane JM, Schneider K, Koza A, Christensen HB, Klein T, Phaneuf PV, Herrgard M, Feist AM, Nielsen AT (2017) Increased production of l-serine in Escherichia coli through adaptive laboratory evolution. Metab Eng 39:141–150. https://doi.org/10.1016/j.ymben.2016.11.008
Munjal N, Mattam AJ, Pramanik D, Srivastava PS, Yazdani SS (2012) Modulation of endogenous pathways enhances bioethanol yield and productivity in Escherichia coli. Microb Cell Fact 11:145. https://doi.org/10.1186/1475-2859-11-145
Ohtake T, Pontrelli S, Lavina WA, Liao JC, Putri SP, Fukusaki E (2017) Metabolomics-driven approach to solving a CoA imbalance for improved 1-butanol production in Escherichia coli. Metab Eng 41:135–143. https://doi.org/10.1016/j.ymben.2017.04.003
Procentese A, Raganati F, Olivieri G, Russo ME, Salatino P, Marzocchella A (2015) Continuous xylose fermentation by Clostridium acetobutylicum—assessment of solventogenic kinetics. Bioresour Technol 192:142–148. https://doi.org/10.1016/j.biortech.2015.05.041
Royce LA, Yoon JM, Chen Y, Rickenbach E, Shanks JV, Jarboe LR (2015) Evolution for exogenous octanoic acid tolerance improves carboxylic acid production and membrane integrity. Metab Eng 29:180–188. https://doi.org/10.1016/j.ymben.2015.03.014
Saini M, Lin LJ, Chiang CJ, Chao YP (2017) Synthetic consortium of Escherichia coli for n-butanol production by fermentation of the glucose–xylose mixture. J Agric Food Chem 65:10040–10047. https://doi.org/10.1021/acs.jafc.7b04275
Shen CR, Lan EI, Dekishima Y, Baez A, Cho KM, Liao JC (2011) Driving forces enable high-titer anaerobic 1-butanol synthesis in Escherichia coli. Appl Environ Microbiol 77:2905–2915. https://doi.org/10.1128/AEM.03034-10
Shen CR, Liao JC (2008) Metabolic engineering of Escherichia coli for 1-butanol and 1-propanol production via the keto-acid pathways. Metab Eng 10:312–320. https://doi.org/10.1016/j.ymben.2008.08.001
Sherkhanov S, Korman TP, Bowie JU (2014) Improving the tolerance of Escherichia coli to medium-chain fatty acid production. Metab Eng 25:1–7. https://doi.org/10.1016/j.ymben.2014.06.003
Tao H, Gonzalez R, Martinez A, Rodriguez M, Ingram LO, Preston JF, Shanmugam KT (2001) Engineering a homo-ethanol pathway in Escherichia coli: increased glycolytic flux and levels of expression of glycolytic genes during xylose fermentation. J Bacteriol 183:2979–2988. https://doi.org/10.1128/JB.183.10.2979-2988.2001
Tian T, Salis HM (2015) A predictive biophysical model of translational coupling to coordinate and control protein expression in bacterial operons. Nucleic Acids Res 43:7137–7151. https://doi.org/10.1093/nar/gkv635
Wiesenborn DP, Rudolph FB, Papoutsakis ET (1988) Thiolase from Clostridium acetobutylicum ATCC 824 and its role in the synthesis of acids and solvents. Appl Environ Microbiol 54:2717–2722
Wu Y, Xue C, Chen L, Yuan W, Bai F (2016) Synergistic effect of calcium and zinc on glucose/xylose utilization and butanol tolerance of Clostridium acetobutylicum. FEMS Microbiol Lett 363:fnw023. https://doi.org/10.1093/femsle/fnw023
Xia PF, Zhang GC, Walker B, Seo SO, Kwak S, Liu JJ, Kim H, Ort DR, Wang SG, Jin YS (2017) Recycling carbon dioxide during xylose fermentation by engineered Saccharomyces cerevisiae. ACS Synth Biol 6:276–283. https://doi.org/10.1021/acssynbio.6b00167
Xue C, Zhao XQ, Liu CG, Chen LJ, Bai FW (2013) Prospective and development of butanol as an advanced biofuel. Biotechnol Adv 31:1575–1584. https://doi.org/10.1016/j.biotechadv.2013.08.004
Zhou P, Zhang Y, Wang P, Xie J, Ye Q (2013) Butanol production from glycerol by recombinant Escherichia coli. Ann Microbiol 64:219–227. https://doi.org/10.1007/s13213-013-0654-5
Zheng J, Tashiro Y, Yoshida T, Gao M, Wang Q, Sonomoto K (2013) Continuous butanol fermentation from xylose with high cell density by cell recycling system. Bioresour Technol 129:360–365. https://doi.org/10.1016/j.biortech.2012.11.066
Acknowledgements
The authors thank Prof. Ramon Gonzalez for providing E. coli LA07 strain. ASA is a recipient of Arturo Falaschi Postdoctoral Fellowship from ICGEB and KJ is a recipient of Research Fellowship from CSIR.
Funding
This study was funded by Department of Biotechnology, Government of India, via Grant no. BT/PR/Centre/03/2011.
Author information
Authors and Affiliations
Contributions
KJ, ASA and SSY designed the study. ASA and KJ executed all the experiments. ASA, KJ and SSY analyzed all the data. SSY, ASA and KJ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Abdelaal, A.S., Jawed, K. & Yazdani, S.S. CRISPR/Cas9-mediated engineering of Escherichia coli for n-butanol production from xylose in defined medium. J Ind Microbiol Biotechnol 46, 965–975 (2019). https://doi.org/10.1007/s10295-019-02180-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-019-02180-8