
Abstract
Lignin valorization can be obtained through cleavage of selected bonds by microbial enzymes, in which lignin is segregated from cellulose and hemicellulose and abundant phenolic compounds can be provided. In this study, Pseudomonas sp. Q18, previously isolated from rotten wood in China, was used to degrade alkali lignin and raw lignocellulosic material. Gel-permeation chromatography, field-emission scanning electron microscope, and GC–MS were combined to investigate the degradation process. The GC–MS results revealed that the quantities of aromatic compounds with phenol ring from lignin increased significantly after incubation with Pseudomonas sp. Q18, which indicated the degradation of lignin. According to the lignin-derived metabolite analysis, it was proposed that a DyP-type peroxidase (PmDyP) might exist in strain Q18. Thereafter, the gene of PmDyP was cloned and expressed, after which the recombinant PmDyP was purified and the enzymatic kinetics of PmDyP were assayed. According to results, PmDyP showed promising characteristics for lignocellulosic biodegradation in biorefinery.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lignocellulosic material is of significant interest due to its potential capability to produce substitutions for petroleum-derived chemicals [39, 48]. Lignin, consisting of phenylpropane units connected by ether and C–C linkages, is a branched polymer of high complexity, and is mainly derived from three monolignols: the 4-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) units, respectively [5, 33, 47]. The β-O-4 aryl ether linkages are the most abundant (accounting for 50–70%) followed by β-β, β-5, 5–5, and 5-O-4 linkages [45].
As part of an efficient biorefinery process, the pretreatment of lignocellulosic material is a key step to break down or loosen highly resistant lignin. Various physical, chemical, and biological approaches have been attempted for this pretreatment [19, 21, 27, 65]. Besides, fungi [23, 51, 66] and bacteria [61] have been studied for use in the degradation of lignin. Although some bacteria have been reported to depolymerize lignin, the enzymology of bacterial lignin degradation is poorly understood until recently. The dye-decolorizing peroxidase (DyP)-type enzymes (EC 1.11.1.19) have been studied for the ability to degrade lignin. The novel peroxidase DypB from the lignolytic soil bacterium Rhodococcus jostii RHA1 was demonstrated to catalyze the peroxide-dependent oxidation of divalent manganese [53]. Three DyPs were found in Pseudomonas fluorescens Pf-5, and Dyp1B enzyme showed oxidation activity for Mn(II) and Kraft lignin [40]. Different bacteria may employ different pathways of lignin degradation [3].
In this study, the ability of Pseudomonas sp. Q18 to degrade native lignocellulosic material and alkali lignin (AL) was investigated. Pseudomonas sp. Q18 was first isolated from rotten wood in Qinling Mountain, Shaanxi province China [63]. The degradation results were used to identify and characterize the DyP-type peroxidase from Pseudomonas sp. Q18.
Materials and methods
Microorganism, culture conditions, and chemicals
Pseudomonas sp. Q18 (Accession No. KX822686) was isolated from rotten wood in the forests of Qinling Mountain in Shaanxi province, China, as described in our previous article [63]. The phylogenetic tree of strain Q18 is shown in Fig. S1. Escherichia coli DH5α and BL21(DE3) (TransGen Biotech Co., Ltd) were used for gene cloning, and expression. The plasmid pET-28a(+) (Takara Bio, Otsu, Japan) was used as the expression vector. Wheat straw, corn stalk, and switchgrass were collected from a farmhouse in Xianyang City of Shaanxi province. All chemicals and biochemical reagents used in this work were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Biodegradation of native lignocellulosic material by strain Q18
Corn stalk, switchgrass, and wheat straw were used as the raw lignocellulosic material. The plant material was pulverized, screened by a 0.38 mm screen, and then dried at 60 °C for 48 h. Strain Q18 was grown in liquid LB medium until reaching an OD 600 of 1.0. Then, they were inoculated (10%, v/v) to the biodegradation medium: 2.5 g of NaNO3, 1.0 g of KH2PO4, 1.0 g of K2HPO4, 1.0 g of MgSO4, 1.0 g of NaCl, 0.5 g of CaCl2, 1.0 g of NH4Cl, and 3.0% (w/v) raw lignocellulosic material in 1000 mL of deionized water (pH 6.0). Strain Q18 was separately inoculated into the medium containing corn stalk, switchgrass, or wheat straw. The cultures were then incubated at 37 °C and 150 rpm for 15 days. Control samples were inoculated without bacteria.
The lignin content in the samples was determined after treatment. The total lignin content included the acid-insoluble lignin (AIL) and acid-soluble lignin (ASL) [36]. The ASL was determined by maximum UV absorbance of the supernatant, after the insoluble lignin was removed, at 205 nm using an extinction coefficient of 110 L g−1 cm−1 [1]. The formula was as follows:
where df is the dilution factor, V is the total volume of the supernatant, A is the UV absorbance, and W is the weight of the initial wood powder. The AIL was determined according to the standard Klason’s method [4, 24]. In a Soxhlet extractor, the samples were extracted with ethanol–benzene (1:1) for 8 h and then dried at 60 °C for 48 h. Next, 72% sulfuric acid was added and mixture was incubated at 15 °C for 2.5 h, before dilution of the sulfuric acid to 3% using deionized water. Subsequently, the samples were autoclaved at 121 °C for 30 min to remove the cellulose and hemicellulose in the residues and determine the lignin content. In this study, the AIL content was calculated by the following formula:
Analysis of field-emission scanning electron microscope (FE-SEM)
Shredded lignocellulosic material degraded by incubation with Pseudomonas sp. Q18 for 15 days were inspected with FE-SEM to visually study the decay processing [28]. To do this, 50 μL medium of the wheat straw sample was placed on a glass slide. The samples were immobilized by the addition of glutaraldehyde (2.5%) for 1 h. Next, a gradient of 30–90% ethanol was used to dehydrate the samples. After the above processing, the samples were coated with a layer of Au/Pd alloy using an ion sputter (HITACHI E-1045) [52]. Samples of AL degraded by strain Q18 for 7 days were collected and centrifuged at 8000×g for 10 min. The supernatant was dried in a vacuum freeze-drying apparatus, before imaging of these samples by FE-SEM (HITACHI S-4800). The controls were cultures lacking bacteria.
Gel-permeation chromatography (GPC) of degradation of alkali lignin
The biodegradation of AL was carried out in medium containing 2.5 g of NaNO3, 1.0 g of KH2PO4, 1.0 g of K2HPO4, 1.0 g of MgSO4, 1.0 g of NaCl, 0.5 g of CaCl2, 1.0 g of NH4Cl, and 3.0 g of alkali lignin in 1000 mL of deionized water (pH 6.0). The strain Q18 was cultured in LB medium until reaching an OD 600 value of 1.0. Then, the flasks containing 50 mL medium were inoculated with bacterial culture (10% v/v), and then, they were incubated at 37 °C, 150 rpm for 7 days. The sample lacking bacteria were analyzed as the control. The molecular weight distribution of AL was determined by GPC [58, 59]. The supernatant of each sample was dried in a vacuum freeze-drying apparatus. Then, a solution of AL in water (20 mg/mL, 5 mL) was loaded onto an Ultrahydrogel™ Linear 300 mm × 7.8 mmid column. The mobile phase was 0.1 mol/L NaNO3 solution with pH 10.7 and the flow rate was 0.5 mL/min. The sample was detected by UV–vis spectroscopy at 254 and 280 nm.
GC–MS analysis of degradation of AL
Samples to assay the amount of AL degraded by Pseudomonas sp. Q18 after 3 and 7 day incubation were prepared as described [7, 20, 25, 32, 37, 42,43,44, 57, 60, 69]. The cultures (50 mL) without (control) and with bacteria were centrifuged at 8000×g for 10 min. The pH of the supernatant was adjusted to 1–2 with the addition of concentrated HCl. After adding ethyl acetate (three volumes), the organic layer of the mixture was collected and dried. Then, the derivatization reaction occurred in the presence of trimethyl silyl [BSTFA (N, O-bis (trimethylsilyl) trifluoroacetamide and TMCS (trimethylchlorosilane)] (Sigma-Aldrich), after heating at 60 °C for 15 min.
The injection volume was set to 1 µL and the samples were applied and separated by GC–MS (GCMS-QP2010 Ultra). The flow rate of helium was 1 mL/min. The column was held at 50 °C for 5 min, increased to 300 °C at a rate of 10 °C/min, and then held for 5 min. The transfer line and ion source temperatures were maintained at 200 and 250 °C. Electron ionization (EI) mass spectra were recorded at 30-550 (m/z) at electron energy of 70 eV. Based on the retention times (RT), the compounds were identified by comparison to the NIST library.
Cloning and expression of pmDyP in Escherichia coli
Pseudomonas sp. Q18 was cultured in LB medium at 37 °C, 150 rpm for 24 h. The cells were obtained by centrifugation at 4 °C, 8000×g, and the genomic DNA was extracted using an Ezup column bacteria genomic DNA purification kit (Shanghai Sangon Biotech Co., Ltd.), from a total of 1 mL of the culture. The chromosomal DNA was used as a template for gene-specific primed PCR amplification. The gene-encoding pmDyP was amplified using primers, designed based on the gene sequence of the hypothetical DyP-type peroxidase of Pseudomonas sp. JY-Q (CP011525.1): pm-F: 5′-AATTCCATGGGCATGCCGTTCCAGCAAGGTCTGCTTGCC, and pm-R: 5′-AATGCTCGAGGGCCCGCAGCAAGGAGCTCAGAT (the restriction sites NcoI and XhoI were underlined). Then, the recombinant plasmid pET28a-pmDyP was transformed into E. coli DH5α. The successful cloning of the pmDyP gene was confirmed by sequencing by Shanghai Sangon Biotech Company (China).
The resulting plasmid with the pmDyP gene under the control of the T7lac promoter was transformed into the host expression strains E. coli BL21(DE3). Expression of PmDyP was induced by addition of IPTG (0.2 mM) when the culture had reached an optical density of about 0.6 at 600 nm. The cells were collected by centrifugation (6000×g for 10 min at 4 °C) and resuspended in 2 mL of 20 mM Tris–HCl buffer (pH 8.0) containing 5 mM NaCl, followed by ultrasonication for 20 min using a SCIENTZ-IID ultrasonic homogenizer (Ningbo Scientz Biotechnology Polytron Technologies Inc., Zhejiang province, China). The resulting cell lysates were centrifuged at 8000×g for 30 min at 4 °C. SDS-PAGE was then performed to analyze the supernatant and the insoluble fraction of the sonicated whole cell lysate [10, 63].
Purification and refolding of PmDyP from inclusion bodies
The precipitate was dissolved in extraction buffer (20 mM Tris–HCl, 150 mM NaCl, 20 mM imidazole with 8 M urea, pH 8.0) overnight at 4 °C. After centrifugation, the supernatant was loaded onto an Ni–NTA His Bind resin column (Novagen, Germany) and the protein of PmDyP was eluted by elution buffer (20 mM Tris–HCl, 150 mM NaCl, 8 M urea, with 250 mM imidazole, pH 8.0). The eluted protein was detected by SDS-PAGE. The eluate containing PmDyP was diluted with extraction buffer to a final protein concentration of 0.5 mg/mL. The samples were dialyzed to refold in renaturation buffer (20 mM Tris–HCl, 150 mM NaCl, 0.5 mM GSH (Glutathione reduced), 0.05 mM GSSG (l-glutathione oxidized), 10 mM glycine, containing different concentrations of urea) at 4 °C with stirring at 120 rpm, and the renaturation buffer was changed every 8 h with decreasing urea concentration. The protein concentration was determined by BCA method with bovine serum albumin as the standard [8, 67].
Bioinformatic and phylogenetic analysis of PmDyP
The plasmid pET28a-pmDyP was sequenced, and the databases of PeroxiBase and NCBI were used to explore the protein homology and conserved motifs of the deduced amino acid sequence of PmDyP obtained from Pseudomonas sp. Q18. The phylogenetic tree analysis of the reported DyP-type A–D protein was constructed using the MEGA 5.0 and iTOL based on the algorithm of neighbor-joining (NJ) [2, 49]. The sequence for the signal peptide, transmembrane regions, and the hydrophobicity of the protein were identified and analyzed by SignalP3.0, TMHMM, TMPRED at the ExPASy server, and ExPASy-ProtScale [15, 34]. Sequence alignments of the reported primary sequences of DyPs were performed using ClustalW Multalign [56, 71]. The deduced tertiary structure of PmDyP was predicted using I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) [64].
Steady-state kinetic assays
All assays were determined at 25 °C in 100 mM phosphate buffer (pH 5.0) using a microplate reader (Victor X3, PerkinElmer, USA). Enzyme activity was determined at different concentrations of the following substrates. The oxidation of ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid)), at concentrations 0.05–10 mM, was performed with 1 mM hydrogen peroxide, with monitoring at 420 nm (ε420 = 36,000 M−1 cm−1). The oxidation of guaiacol, at concentrations 0.05–5 mM, was studied with 1 mM hydrogen peroxide, with monitoring at 470 nm (ε470 = 26,600 M−1 cm−1). The oxidation of 2,6-DMP (2,6-Dimethylphenol), at concentrations 0.05–5 mM, was performed with 1 mM hydrogen peroxide, with monitoring at 468 nm (ε468 = 49,600 M−1 cm−1). The oxidation of MnCl2, at concentrations 0.05–25 mM, was performed with 1 mM hydrogen peroxide, with monitoring at 238 nm (ε468 = 6500 M−1 cm−1). Steady-state kinetic parameters were measured by non-linear curve fitting to the obtained enzyme activities (using Graphpad Prism 5 software) and were fitting to the Michaelis–Menten equation [6, 40, 56].
Statistical analysis
All experiments were performed in triplicate, and the results were expressed as mean ± standard deviation. Data analysis was performed by ANOVA and Duncan test using SPSS 16.0. In all analysis, p < 0.05 was considered significant.
Results and discussion
Biodegradation of native lignocellulosic material by strain Q18
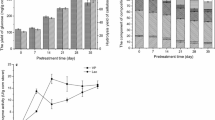
The degradation results of the raw lignocellulosic material are shown in Fig. 1. The measured AIL amounts of native corn stalk, wheat straw, and switchgrass were (21.61 ± 0.57) %, (22.72 ± 0.49) %, and (18.95 ± 0.52) %, respectively. The ASL content of the three kinds of plants was less than 4%. After treatment by Pseudomonas sp. Q18, the amount of lignin remaining decreased. Most ASL was degraded, which was agreed with a previous report [12]. The amount of the remaining material compared with the total lignin was lower for switchgrass (14.60 ± 0.79) % than it was for corn stalk and wheat straw. The AIL content was also the lowest, which indicated that the Pseudomonas sp. Q18 exhibited the best degradation capability on switchgrass. The diversity of plant biomass types results in differences in composition and structure among these feedstocks, which plays an important role in process of the plant biomass conversion. Plant biomass consists of lignin, carbohydrate polymers, pectins, proteins, and other components [13]. Grass lignin is composed of all G, S, and H sub-units which differs from the composition present in hardwoods (G and S) and softwoods (G and H) [30, 50]. The S/G ratio can influence structural features, such as the amount of β-O-4 linkages, methoxyl content, or the degree of condensation [50]. Compared with wheat straw and corn stalk, the treatment of switchgrass by Pseudomonas sp. Q18 showed the highest weight loss of dry biomass, almost 25%. The degradation results of the lignin in native lignocellulosic material indicated that Pseudomonas sp. Q18 had high potential to break down lignin [11, 26]. Compared with Rhizobia sp. YS-1r, Pseudomonas sp. Q18 had great potential capacity for raw lignin degradation. Rhizobia sp. YS-1r was reported to degrade 15% of AIL in switchgrass [22], much less than the 25% observed here. However, the activity of Pseudomonas sp. Q18 was lower (about 20%) than the reported activity of the fungi, such as Ganoderma applanatum BEOFB 411 with a maximum rate of wheat straw lignin degradation of 35% during cultivation for 14 days [14]. The morphological changed in wheat straw after treatment by strain Q18 were observed by FE-SEM. There were obvious changes before and after degradation. The control wheat straw seemed smooth and plump on the surface. After 15 day microbial treatment, the structure was disrupted and many small fragments were attached to the surface.

Changes of total lignin content and loss weight after pretreatment by Pseudomonas sp. Q18 for 15 days. In addition, the images of degraded wheat straw treated by strain Q18 by FE-SEM. Control samples were inoculated without bacteria. AIL acid-insoluble lignin, ASL acid-soluble lignin, LW loss weight of dry native straw. Total lignin (%) = AIL content (%) + ASL (%). All experiments were performed in triplicate, and the results were expressed as mean ± standard deviation
Degradation of AL by strain Q18
GPC was used to measure the molecular weight distribution of AL before and after the bacterial degradation by Pseudomonas sp. Q18. The number-average (Mn), weight-average (Mw) molecular weights, and polydispersity (Mw/Mn) are shown in Fig. 2 and Table 1. The AL was used, as a bacteria-free control, and showed a molecular weight (Mw) range from 53 to 68,599 Da. The Mw of the control sample indicated that the AL included both high and low Mw lignin fractions. The fraction with fragment of Mw greater than 2000 Da in size was about 20%. After 7 days of Pseudomonas sp. Q18 treatment, the highest Mw was decreased to 48772 Da, and only 10% of fragments had the Mw more than 1500 Da. This result indicated that the molecular weight tended to be low values after treatment by bacteria, which was coincidence with the lignin content after treatment. The GPC results indicated that the AL particles with high Mw were depolymerized into smaller ones after treatment. In addition, the analysis of untreated and treated AL by FE-SEM showed that the control sample maintained irregular spherical shapes with porous internal structure, and the surface was mostly smooth, although some breakage might have occurred during processing. The diameter of these AL particles was greater than 100 μm. However, the smooth surface of the lignin was completely eroded after microbial treatment. The Cupriavidus basilensis B-8 also could break down the kraft lignin from 15.1 to 1.65 kDa for 7 days [52]. For these processes, the degraded lignin fraction could be consumed as the carbon source for bacteria metabolism.

GPC chromatograms of molecular weight distribution and FE-SEM images of changes in the control sample and AL treated by strain Q18. The sample lacking bacteria were analyzed as the control
GC–MS analysis of degraded AL
GC–MS was used to analyze the AL degradation compounds with low molecular weight. After treatment with Pseudomonas sp. Q18, the appearance of new peaks and reduction of peaks indicated the generation of new metabolic products [26]. The numbers of peaks changed (Fig. S2), and the aromatic compounds identified from the peaks of degradation for 0 (control), 3, and 7 days are shown in Table 2. There were 12, 9, and 17 aromatic compounds detected in the control, 3 and 7 day samples, respectively.
According to Table 2 and Fig. 3, the high molecular weight compounds, i.e., Compound 25 (1,2-benzenedicarboxylic acid, dinonyl ester) (RT = 29.689 min) and Compound 26 (1,2-benzenedicarboxylic acid, ditridecyl ester) (RT = 29.935 min), detected in all samples, were the main compounds in the control sample. Additionally, in the control sample, a small proportion of aromatic monomer compounds were found, like Compounds 12, 14, 20, and 28. These compounds were considered to be the basic units that built the lignin structure, and may have derived from the industrial process.

Proportion of absorption peaks area of aromatic compounds in the control (0 day) (a), third day (b), and seventh day (c) samples treated by strain Q18
In the 3 day sample, the complex aromatic compounds, Compound 16 (17.94%), Compound 24 (12.72%), and Compound 25 (8.84%), were decreased compared with the control. However, the relative content of Compound 13 (30.08%) was significantly increased. Most of the low molecular weight aromatic monomers (i.e., Compounds 12 and 14) disappeared. The results were the same as those observed in the previous literature of co-culture of Bacillus subtilis and Klebsiella pneumonia for Kraft lignin degradation [62]. It indicated that lignin was degraded by strain Q18. Meanwhile, the massive consumption and catabolism of aromatic compounds showed that the strain Q18 had the ability to utilize the low molecular weight compounds as a carbon or energy source [48, 62, 70]. Lignin depolymerization, aromatic catabolism, and co-product generation occurred simultaneously.
Lignin is a branched polymer of high complexity that consists of substituted phenylpropane units joined by C–C and ether linkages [5, 33, 47]. After incubation for 7 days (Fig. 3c), the relative content of these high molecular weight compounds (Compounds 16 and 24) was still decreased and some typical lignin degradation compounds were identified (Table 2). Compared with the control, a significant increase in the number of peaks was found after 7 day treatment, which was consistent with other studies [16, 57]. 1-(4-hydroxy-3,5-dimethoxyphenyl)-ethanone (Compound 22, S unit), a typical lignin monomer unit, was found in the 7 day sample and its relative content accounted for 18.51%, as well as Compound 15 (2.56%). The G unit monomer, 1-(4-hydroxy-3-methoxyphenyl)-ethanone (Compound 18, 4.05%) and 2-methoxy-4-vinylphenol (Compound 14, 1.39%) appeared as degraded products in the cultures.
In the degradation process, the amounts of organic acids and esters increased, such as oxalic acid, ethyl acetate, and 3-acetyloxybutanoic acid ethyl ester [9, 31]. This increase possibly reflects chemical reaction among microbial primary metabolites or the intermediate cleavage of lignin after degradation.
It was reported that more than 160 kinds of single phenyl ring compounds could be obtained from lignin, such as p-coumaryl alcohol, coniferyl alcohol, sinapyl alcohol, and their derivatives [68]. However, in this study and previous literatures, less than 20 single phenyl ring aromatic products were identified, because more compounds might be in very small amounts outside the GC–MS scale [70]. Benzenepropanoic acid, syringic acid, and ferulic acid were found in kraft lignin degraded by Acetoanaerobium sp. WJDL-Y2 detected by GC–MS [16]. Some aromatic compounds like vanilic acid, coniferyl, and sinapyl alcohols were important intermediates or products, and their presence in the alkali lignin degradation production could be related to the oxidation of lignin [38, 44, 46, 57]. In a recent study, vanillic acid was determined to be a highly abundant aromatic metabolite (44.2%) of alkali lignin after 5 days of treatment by Bacillus ligniniphilus L1 [70].
Bioinformatic analysis of PmDyP
Combined with the analysis of aromatic compounds and the reported metabolic pathways [70], a DyP-type peroxidase was thought to be present in Pseudomonas sp. Q18. The primers of PmDyP were designed according to the sequence of the hypothetical protein of DyP-type peroxidase reported in NCBI, especially the sequences of related Pseudomonas sp. strains. The results of PCR amplification showed that PmDyP could be amplified using the primers designed according to the sequences of the DyP genes of Pseudomonas sp. JY-Q. Then, sequence of PmDyP was cloned into pET-28a(+) and transformed into E. coli DH5α. Analysis of the nucleotide sequence of PmDyP indicated that the cloned DNA contained a single open reading frame of 864 bp which encoded a protein monomer of 287 amino acids with a deduced theoretical pI of 4.71 [18, 29]. The cloned enzyme contained a signature DyP-type peroxidase motif after analysis of protein domain databases (Fig. S3).
According to the previous literature, DyPs were divided into four (A–D) subfamilies with bacterial and fungal enzymes phylogenetically [35, 49]. With the construction of a phylogenetic tree and the maximum likelihood analysis of reported DyPs (Fig. 4), PmDyP was found to be closest to PpDyP (GI: 501229266), which was reported to belong to the B-type subfamily [49]. However, 20 amino acids were different in these proteins, which may lead to the different activity. Most homology analysis has focused on primary, secondary and tertiary structure homology. Some homologous proteins have been analyzed, though most of the proteins remain theoretical or their function is unknown. This study chose two well-studied and shared proteins (DyPPa and TyrA) that are homologous to PmDyP in primary structure. Compared with the reported primary sequences of DyPs, PmDyP shared highly conserved amino acid residues (Fig. 5), such as Asp132 (D132), His197 (H197), Arg214 (R214), and the GXXDG motif which played a significant role in the formation of the catalytic centers and the functions of the DyP-type peroxidase family. PmDyP was identified with 48.67 and 60.88% amino acid homology to DyPPa and TyrA, respectively. Like the tertiary structure of DyPPa and TyrA, PmDyP showed a typical spatial structure. The predicted structure of PmDyP includes two domains, four-stranded antiparallel β-sheets and peripheral α-helices [29, 55]. The structural characteristics supported the hypothesis that PmDyP was a member of the DyP-type peroxidase family. As reported previously, D132, H197, R214, and GXXDG motifs were thought to be related to the heme peroxidase catalytic cycle process and form the activity center. The bent degree of R242 and R245 in TyrA and BtDyP, respectively, lead to different activities, suggesting that the state of R214 might influence enzyme activity [17, 49, 54].

Radial phylogram of DyPs. The names of bacterial or fungal strains are indicated in subscript with the protein or abbreviated organism names. Phylogenetic tree comparing PmDyP (bold) with isolated DyPs belonging to subfamilies A–D: DyP A: TfuDyp from Thermobifida fusca (GI: 71917209), Mva from Mycobacterium vanbaalenii PYR-1 (ABM12972.1), Bsu168 from Bacillus subtilis 168 (CAB15852.1), Pde from Paracoccus denitrificans PD1222 (ABL69832.1), Rpa from Rhodopseudomonas palustris BisB18 (ABD87513.1). DyP B: DyPPa from Pseudomonas aeruginosa (GI: 94829180), CteKF-1 from Comamonas testosteroni KF-1 (EED66859.1), PpDyP from Pseudomonas putida (GI: 501229266), YfeX from E. coli K-12 (BAE76711.1), Ddi from Dictyostelium discoideum AX4 (EAL70759.1), Acsp from Acinetobacter sp. ADP1 (CAG67144.1), RjoDypB from Rhodococcus jostii (GI: 330689635), BtDyP from Bacteroides thetaiotaomicron VPI-5482 (2GVK), Cco from Campylobacter concisus 13826 (EAT98288.1). DyP C: Chu from Cytophaga hutchinsonii ATCC 49185 (ABG59511.1), Cysp from Cyanothece sp. PCC 7424 (ACK71272.1), Cvi from Chromobacterium violaceum ATCC 12472 (AAQ59612.1), Oan from Ochrobactrum anthropi ATCC 49185 (ABS17389.1), Pssp from Psychrobacter sp. PRwf-1 (ABQ94167.1), MxDyP from Myxococcus xanthus (GI: 108465542), SavDyP from Streptomyces avermitilis (GI: 29604188), Ana from Anabaena sp. PCC 7120 (BAB77951.1), AmspDyp2 from Amycolatopsis sp. Atcc 39116 (4G2C_B). DyP D: Lbi2 from Laccaria bicolor S238 N-H82 (EDR12662.1), Ppl from Postia placenta Mad-698-R (EED79944.1), Pch from Penicillium chrysogenum Wisconson 54-1255 (CAP99029.1), BjaDyP from Bjerkandera adusta Dec1 (GI: 4760440), AauDyP1 from Auricularia auricula-judae (GI: 433286646), Pos from Pleurotus ostreatus (CAK55151.1), Msp1 from Marasmius scorodonius (GI: 261266601)

Deduced tertiary structure of PmDyP using I-TASSER with the C-score of 1.16. In addition, the multiple alignment of amino acid sequences for PmDyP, DyPPa, and TyrA. DyPPa from Pseudomonas aeruginosa PKE117 (GU967380), TyrA from Shewanella oneidensis (NP716371)
Expression and steady-state kinetic characterization
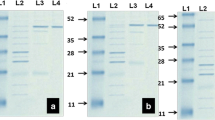
The PmDyP was expressed and purified. As shown in Fig. S4, the PmDyP was expressed as insoluble inclusion bodies. The inclusion bodies were dissolved in denaturation buffer and purified by the Ni–NTA agarose column. After the refolding process, the purified PmDyP was analyzed by SDS-PAGE (Fig. 6), which showed that its molecular weight was about 31.7 kDa. Steady-state kinetic characterization was performed using a range of peroxidase substrates, providing the Km and kcat values, as listed in Table 3 (original data shown in Supporting information, Fig. S5). PmDyP showed catalytic activity with all tested peroxidase substrates. However, according to the previous report, there was no activity to be observed with Mn2+ and guaiacol for TfuDyP. The kcat/Km value of PmDyP was 7000 using ABTS as the substrate, which was higher than the value of TfuDyP [41]. In addition, for guaiacol and Mn2+, PmDyP showed higher values of kcat/Km than Dyp1B, Dyp2B, and DyPA from P. fluorescens Pf-5 [40]. The steady-state kinetic characterization demonstrated the potential peroxide ability of PmDyP for biotechnological applications.

Result of SDS-PAGE analysis of protein. Lane 1, insoluble fraction of sonicated whole cell lysate after 6 h induction at 30 °C with 0.2 mM IPTG; lane 2, soluble fraction of sonicated whole cell lysate after 6 h induction at 30 °C with 0.2 mM IPTG; lane 3, inclusion bodies dissolved in extraction buffer; lane 4, eluate with 250 mM imidazole; M, protein molecular weight marker
Conclusion
In summary, Pseudomonas sp. Q18 was proved to have the excellent ability to break down alkaline lignin and native lignocellulosic material. Many kinds of relative aromatic compounds were identified as changing in the process of lignin degradation. Combined with the reported metabolic pathways and the results in this paper, a novel DyP-type peroxidase, PmDyP, was cloned and expressed from Pseudomonas sp. Q18. Steady-state kinetic characterization suggested that PmDyP was a potential peroxidase. Future work should explore more relative metabolites and enzyme characterization combined with new technologies such as genetic engineering and bioinformatics.
References
Abe M, Yamanaka S, Yamada H, Yamada T, Ohno H (2015) Almost complete dissolution of woody biomass with tetra-n-butylphosphonium hydroxide aqueous solution at 60 °C. Green Chem 17:4432–4438. https://doi.org/10.1039/c5gc00646e
Ahmad M, Roberts JN, Hardiman EM, Singh R, Eltis LD, Bugg TD (2011) Identification of DypB from Rhodococcus jostii RHA1 as a lignin peroxidase. Biochemistry 50:5096–5107. https://doi.org/10.1021/bi101892z
Ahmad M, Taylor CR, Pink D, Burton K, Eastwood D, Bending GD, Bugg TD (2010) Development of novel assays for lignin degradation: comparative analysis of bacterial and fungal lignin degraders. Mol BioSyst 6:815. https://doi.org/10.1039/b908966g
Azizi-Shotorkhoft A, Mohammadabadi T, Motamedi H, Chaji M, Fazaeli H (2016) Isolation and identification of termite gut symbiotic bacteria with lignocellulose-degrading potential, and their effects on the nutritive value for ruminants of some by-products. Anim Feed Sci Technol 221:234–242. https://doi.org/10.1016/j.anifeedsci.2016.04.016
Boerjan W, Ralph J, Baucher M (2003) Lignin biosynthesis. Annu Rev Plant Biol 54:519–546. https://doi.org/10.1146/annurev.arplant.54.031902.134938
Brissos V, Tavares D, Sousa AC, Robalo MP, Martins LO (2017) Engineering a bacterial Dyp-type peroxidase for enhanced oxidation of lignin-related phenolics at alkaline pH. ACS Catal 7:3454–3465. https://doi.org/10.1021/acscatal.6b03331
Brzonova I, Kozliak E, Kubátová A, Chebeir M, Qin W, Christopher L, Ji Y (2014) Kenaf biomass biodecomposition by basidiomycetes and actinobacteria in submerged fermentation for production of carbohydrates and phenolic compounds. Bioresour Technol 173:352–360. https://doi.org/10.1016/j.biortech.2014.09.057
Bu D, Zhou Y, Tang J, Jing F, Zhang W (2013) Expression and purification of a novel therapeutic single-chain variable fragment antibody against BNP from inclusion bodies of Escherichia coli. Protein Expression Purif 92:203–207. https://doi.org/10.1016/j.pep.2013.10.002
Bugg TD, Ahmad M, Hardiman EM, Rahmanpour R (2011) Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep 28:1883–1896. https://doi.org/10.1039/c1np00042j
Camarero S, Ibarra D, Martínez MJ, Martínez ÁT (2005) Lignin-derived compounds as efficient laccase mediators for decolorization of different types of recalcitrant dyes. Appl Environ Microbiol 71:1775–1784. https://doi.org/10.1128/aem.71.4.1775-1784.2005
Chandra R, Raj A, Purohit H, Kapley A (2007) Characterisation and optimisation of three potential aerobic bacterial strains for kraft lignin degradation from pulp paper waste. Chemosphere 67:839–846. https://doi.org/10.1016/j.chemosphere.2006.10.011
Chang YC, Choi D, Takamizawa K, Kikuchi S (2014) Isolation of Bacillus sp. strains capable of decomposing alkali lignin and their application in combination with lactic acid bacteria for enhancing cellulase performance. Bioresour Technol 152:429–436. https://doi.org/10.1016/j.biortech.2013.11.032
Chen HZ, Liu ZH, Qu Y, Wu WT (2015) Steam explosion and its combinatorial pretreatment refining technology of plant biomass to bio-based products. Biotechnol J 10:866. https://doi.org/10.1002/biot.201400705
Ćilerdžić J, Stajić M, Vukojević J (2016) Degradation of wheat straw and oak sawdust by Ganoderma applanatum. Int Biodeterior Biodegrad 114:39–44. https://doi.org/10.1016/j.ibiod.2016.05.024
Cserzö M, Wallin E, Simon I, Von HG, Elofsson A (1997) Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Eng 10:673–676. https://doi.org/10.1093/protein/10.6.673
Duan J, Huo X, Du W, Liang J, Wang D, Yang S (2016) Biodegradation of kraft lignin by a newly isolated anaerobic bacterial strain, Acetoanaerobium sp. WJDL-Y2. Lett Appl Microbiol 62:55–62. https://doi.org/10.1111/lam.12508
Dunford HB (1999) Heme peroxidases. Encycl Biophys. https://doi.org/10.1007/978-3-642-16712-6_52
Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FS (2005) PSORTb v. 2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics 21:617–623. https://doi.org/10.1093/bioinformatics/bti057
Gilca IA, Ghitescu RE, Puitel AC, Popa VI (2014) Preparation of lignin nanoparticles by chemical modification. Iran Polym J 23:355–363. https://doi.org/10.1007/s13726-014-0232-0
Haq I, Kumar S, Kumari V, Singh SK, Raj A (2016) Evaluation of bioremediation potentiality of ligninolytic Serratia liquefaciens for detoxification of pulp and paper mill effluent. J Hazard Mater 305:190–199. https://doi.org/10.1016/j.jhazmat.2015.11.046
Huang XF, Santhanam N, Badri DV, Hunter WJ, Manter DK, Decker SR, Vivanco JM, Reardon KF (2013) Isolation and characterization of lignin-degrading bacteria from rainforest soils. Biotechnol Bioeng 110:1616–1626. https://doi.org/10.1002/bit.24833
Jackson CA, Couger MB, Prabhakaran M, Ramachandriya KD, Canaan P, Fathepure BZ (2017) Isolation and characterization of Rhizobium sp. strain YS-1r that degrades lignin in plant biomass. J Appl Microbiol 122:940–952. https://doi.org/10.1111/jam.13401
Karim M, Daryaei MG, Torkaman J, Oladi R, Ghanbary MAT, Bari E (2016) In vivo investigation of chemical alteration in oak wood decayed by Pleurotus ostreatus. Int Biodeterior Biodegrad 108:127–132. https://doi.org/10.1016/j.ibiod.2015.12.012
Kirk TK, Obst JR (1988) Lignin determination. Methods Enzymol 161:87–101. https://doi.org/10.1016/0076-6879(88)61014-7
Ko JJ, Shimizu Y, Ikeda K, Kim SK, Park CH, Matsui S (2009) Biodegradation of high molecular weight lignin under sulfate reducing conditions: lignin degradability and degradation by-products. Bioresour Technol 100:1622–1627. https://doi.org/10.1016/j.biortech.2008.09.029
Kumar M, Singh J, Singh MK, Singhal A, Thakur IS (2015) Investigating the degradation process of kraft lignin by β-proteobacterium, Pandoraea sp.ISTKB. Environ Sci Pollut Res 22:15690–15702. https://doi.org/10.1007/s11356-015-4771-5
Kumar P, Barrett DM, Delwiche MJ, Stroeve P (2009) Methods for pretreatment of lignocellulosic biomass for efficient hydrolysis and biofuel production. Ind Eng Chem Res 48:3713–3729. https://doi.org/10.1021/ie801542g
Kuuskeri J, Häkkinen M, Laine P, Smolander OP, Tamene F, Miettinen S, Nousiainen P, Kemell M, Auvinen P, Lundell T (2016) Time-scale dynamics of proteome and transcriptome of the white-rot fungus Phlebia radiata: growth on spruce wood and decay effect on lignocellulose. Biotechnol Biofuels 9:192. https://doi.org/10.1186/s13068-016-0608-9
Li J, Liu C, Li B, Yuan H, Yang J, Zheng B (2012) Identification and molecular characterization of a novel DyP-type peroxidase from Pseudomonas aeruginosa PKE117. Appl Biochem Biotechnol 166:774–785. https://doi.org/10.1007/s12010-011-9466-x
Mann DGJ, Labbé N, Sykes RW, Gracom K, Kline L, Swamidoss IM, Burris JN, Davis M, Stewart CN Jr, Haigler C (2009) Rapid assessment of lignin content and structure in switchgrass (Panicum virgatum L.) grown under different environmental conditions. Bioenergy Res 2:246–256. https://doi.org/10.1007/s12155-009-9054-x
Masai E, Katayama Y, Fukuda M (2007) Genetic and biochemical investigations on bacterial catabolic pathways for lignin-derived aromatic compounds. Biosci Biotechnol Biochem 71:1–15. https://doi.org/10.1271/bbb.60437
Mathews SL, Pawlak JJ, Grunden AM (2014) Isolation of Paenibacillus glucanolyticus from pulp mill sources with potential to deconstruct pulping waste. Bioresour Technol 164:100–105. https://doi.org/10.1016/j.biortech.2014.04.093
Mnich E, Vanholme R, Oyarce P, Liu S, Lu F, Goeminne G, Jørgensen B, Motawie MS, Boerjan W, Ralph J (2016) Degradation of lignin β-aryl ether units in Arabidopsis thaliana expressing LigD, LigF and LigG from Sphingomonas paucimobilis SYK-6. Plant Biotechnol J 15:581–593. https://doi.org/10.1111/pbi.12655
Nielsen H, Engelbrecht J, Brunak S, Von HG (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng 10:1–6. https://doi.org/10.1093/protein/10.1.1
Ogola H, Kamiike T, Hashimoto N, Ashida H, Ishikawa T, Shibata H, Sawa Y (2009) Molecular characterization of a novel peroxidase from the cyanobacterium Anabaena sp. strain PCC 7120. Appl Environ Microbiol 75:7509. https://doi.org/10.1128/aem.01121-09
Pamidipati S, Ahmed A (2017) Degradation of lignin in agricultural residues by locally isolated fungus Neurospora discreta. Appl Biochem Biotechnol 181:1561–1572. https://doi.org/10.1007/s12010-016-2302-6
Pandey MP, Kim CS (2011) Lignin depolymerization and conversion: a review of thermochemical methods. Chem Eng Technol 34:29–41. https://doi.org/10.1002/ceat.201000270
Pollegioni L, Tonin F, Rosini E (2015) Lignin-degrading enzymes. FEBS J 282:1190–1213. https://doi.org/10.1111/febs.13224
Ragauskas AJ, Beckham GT, Biddy MJ, Chandra R, Chen F, Davis MF, Davison BH, Dixon RA, Gilna P, Keller M (2014) Lignin valorization: improving lignin processing in the biorefinery. Science 344:1246843. https://doi.org/10.1126/science.1246843
Rahmanpour R, Bugg TD (2015) Characterisation of Dyp-type peroxidases from Pseudomonas fluorescens Pf-5: oxidation of Mn(II) and polymeric lignin by Dyp1B. Arch Biochem Biophys 574:93–98. https://doi.org/10.1016/j.abb.2014.12.022
Rahmanpour R, Rea D, Jamshidi S, Fulop V, Bugg TD (2016) Structure of Thermobifida fusca DyP-type peroxidase and activity towards Kraft lignin and lignin model compounds. Arch Biochem Biophys 594:54–60. https://doi.org/10.2210/pdb5fw4/pdb
Raj A, Chandra R, Reddy MMK, Purohit HJ, Kapley A (2007) Biodegradation of kraft lignin by a newly isolated bacterial strain, Aneurinibacillus aneurinilyticus from the sludge of a pulp paper mill. World J Microbiol Biotechnol 23:793–799. https://doi.org/10.1007/s11274-006-9299-x
Raj A, Kumar S, Haq I, Singh SK (2014) Bioremediation and toxicity reduction in pulp and paper mill effluent by newly isolated ligninolytic Paenibacillus sp. Ecol Eng 71:355–362. https://doi.org/10.1016/j.ecoleng.2014.07.002
Raj A, Reddy MK, Chandra R (2007) Identification of low molecular weight aromatic compounds by gas chromatography–mass spectrometry (GC-MS) from kraft lignin degradation by three Bacillus sp. Int Biodeterior Biodegrad 59:292–296. https://doi.org/10.1016/j.ibiod.2006.09.006
Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, Marita JM, Hatfield RD, Ralph SA, Christensen JH (2004) Lignins: natural polymers from oxidative coupling of 4-hydroxyphenyl-propanoids. Phytochem Rev 3:29–60. https://doi.org/10.1023/b:phyt.0000047809.65444.a4
Rashid GM, Taylor CR, Liu Y, Zhang X, Rea D, Fülöp V, Bugg TD (2015) Identification of manganese superoxide dismutase from Sphingobacterium sp. T2 as a novel bacterial enzyme for lignin oxidation. ACS Chem Biol 10:2286–2294. https://doi.org/10.1021/acschembio.5b00298
Reiter J, Strittmatter H, Wiemann LO, Schieder D, Sieber V (2013) Enzymatic cleavage of lignin β-O-4 aryl ether bonds via net internal hydrogen transfer. Green Chem 15:1373–1381. https://doi.org/10.1039/c3gc40295a
Salvachúa D, Karp EM, Nimlos CT, Vardon DR, Beckham GT (2015) Towards lignin consolidated bioprocessing: simultaneous lignin depolymerization and product generation by bacteria. Green Chem 17:4951–4967. https://doi.org/10.1039/c5gc01165e
Santos A, Mendes S, Brissos V, Martins LO (2013) New dye-decolorizing peroxidases from Bacillus subtilis and Pseudomonas putida MET94: towards biotechnological applications. Appl Microbiol Biotechnol 98:2053–2065. https://doi.org/10.1007/s00253-013-5041-4
Santos RB, Capanema EA, Balakshin MY, Chang H, Jameel H (2012) Lignin structural variation in hardwood species. J Agric Food Chem 60:4923. https://doi.org/10.1021/jf301276a
Schmidt O, Bahmani M, Koch G, Potsch T, Brandt K (2016) Study of the fungal decay of oil palm wood using TEM and UV techniques. Int Biodeterior Biodegrad 111:37–44. https://doi.org/10.1016/j.ibiod.2016.04.014
Shi Y, Yan X, Li Q, Wang X, Xie S, Chai L, Yuan J (2017) Directed bioconversion of Kraft lignin to polyhydroxyalkanoate by Cupriavidus basilensis B-8 without any pretreatment. Process Biochem 52:238–242. https://doi.org/10.1016/j.procbio.2016.10.004
Singh R, Grigg JC, Qin W, Kadla JF, Murphy ME, Eltis LD (2013) Improved manganese-oxidizing activity of DypB, a peroxidase from a lignolytic bacterium. ACS Chem Biol 8:700–706. https://doi.org/10.1021/cb300608x
Sugano Y (2009) DyP-type peroxidases comprise a novel heme peroxidase family. Cell Mol Life Sci 66:1387. https://doi.org/10.1007/s00018-008-8651-8
Sugano Y, Muramatsu R, Ichiyanagi A, Sato T, Shoda M (2007) DyP, a unique dye-decolorizing peroxidase, represents a novel heme peroxidase family: ASP171 replaces the distal histidine of classical peroxidases. J Biol Chem 282:36652–36658
Sugawara K, Nishihashi Y, Narioka T, Yoshida T, Morita M, Sugano Y (2017) Characterization of a novel DyP-type peroxidase from Streptomyces avermitilis. J Biosci Bioeng 123:425–430. https://doi.org/10.1016/j.jbiosc.2016.12.001
Suman SK, Dhawaria M, Tripathi D, Raturi V, Adhikari DK, Kanaujia PK (2016) Investigation of lignin biodegradation by Trabulsiella sp. isolated from termite gut. Int Biodeterior Biodegrad 112:12–17. https://doi.org/10.1016/j.ibiod.2016.04.036
Sun Y, Qiu X, Liu Y (2013) Chemical reactivity of alkali lignin modified with laccase. Biomass Bioenergy 55:198–204. https://doi.org/10.1016/j.biombioe.2013.02.006
Taylor CR, Hardiman E, Ahmad M, Sainsbury P, Norris P, Bugg TD (2012) Isolation of bacterial strains able to metabolize lignin from screening of environmental samples. J Appl Microbiol 113:521–530. https://doi.org/10.1111/j.1365-2672.2012.05352.x
Wang B, Rezenom YH, Cho KC, Tran JL, Lee DG, Russell DH, Gill JJ, Young R, Chu KH (2014) Cultivation of lipid-producing bacteria with lignocellulosic biomass: effects of inhibitory compounds of lignocellulosic hydrolysates. Bioresour Technol 161:162–170. https://doi.org/10.1016/j.biortech.2014.02.133
Wang Y, Liu Q, Yan L, Gao Y, Wang Y, Wang W (2013) A novel lignin degradation bacterial consortium for efficient pulping. Bioresour Technol 139:113–119. https://doi.org/10.1016/j.biortech.2013.04.033
Yadav S, Chandra R (2015) Syntrophic co-culture of Bacillus subtilis and Klebsiella pneumonia for degradation of kraft lignin discharged from rayon grade pulp industry. J Environ Sci 33:229–238. https://doi.org/10.1016/j.jes.2015.01.018
Yang CX, Wang T, Gao LN, Yin HJ, Lu X (2017) Isolation, identification and characterization of lignin-degrading bacteria from Qinling, China. J Appl Microbiol 123:1447–1460. https://doi.org/10.1111/jam.13562
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12:7–8. https://doi.org/10.1038/nmeth.3213
Yang S, Yuan TQ, Li MF, Sun RC (2015) Hydrothermal degradation of lignin: products analysis for phenol formaldehyde adhesive synthesis. Int J Biol Macromol 72:54–62. https://doi.org/10.1016/j.ijbiomac.2014.07.048
Yang Y, Zhou J, Lu H, Yuan Y, Zhao L (2011) Isolation and characterization of a fungus Aspergillus sp. strain F-3 capable of degrading alkali lignin. Biodegradation 22:1017–1027. https://doi.org/10.1007/s10532-011-9460-6
Ye W, Liu J, Wang H, Wang J, Wang X (2012) Cloning, expression, purification, and characterization of a glutamate-specific endopeptidase from Bacillus licheniformis. Protein Expr Purif 82:138–143. https://doi.org/10.1016/j.pep.2011.12.001
Zakzeski J, Bruijnincx PC, Jongerius AL, Weckhuysen BM (2010) The catalytic valorization of lignin for the production of renewable chemicals. Chem Rev 110:3552–3599. https://doi.org/10.1021/cr900354u
Zheng Y, Chai L, Yang Z, Chen Y, Shi Y, Wang Y (2014) Environmentally safe treatment of black liquor with Comamonas sp. B-9 under high-alkaline conditions. J Basic Microbiol 54:152–161. https://doi.org/10.1002/jobm.201200340
Zhu D, Zhang P, Xie C, Zhang W, Sun J, Qian WJ, Yang B (2017) Biodegradation of alkaline lignin by Bacillus ligniniphilus L1. Biotechnol Biofuels 10:44. https://doi.org/10.1186/s13068-017-0735-y
Zubieta C, Krishna SS, Kapoor M, Kozbial P, McMullan D, Axelrod HL, Miller MD, Abdubek P, Ambing E, Astakhova T, Carlton D, Chiu HJ, Clayton T, Deller MC, Duan L, Elsliger MA, Feuerhelm J, Grzechnik SK, Hale J, Hampton E, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kumar A, Marciano D, Morse AT, Nigoghossian E, Okach L, Oommachen S, Reyes R, Rife CL, Schimmel P, van den Bedem H, Weekes D, White A, Xu Q, Hodgson KO, Wooley J, Deacon AM, Godzik A, Lesley SA, Wilson IA (2007) Crystal structures of two novel dye-decolorizing peroxidases reveal a beta-barrel fold with a conserved heme-binding motif. Proteins 69:223–233. https://doi.org/10.1002/prot.21550
Acknowledgements
The authors thank the financial support of Special Fund for Agro-scientific Research in the Public Interest (Grant no. 201503135).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yang, C., Yue, F., Cui, Y. et al. Biodegradation of lignin by Pseudomonas sp. Q18 and the characterization of a novel bacterial DyP-type peroxidase. J Ind Microbiol Biotechnol 45, 913–927 (2018). https://doi.org/10.1007/s10295-018-2064-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-018-2064-y