
Abstract
Yarrowia lipolytica is categorized as a generally recognized as safe (GRAS) organism and is a heavily documented, unconventional yeast that has been widely incorporated into multiple industrial fields to produce valuable biochemicals. This study describes the construction of a CRISPR-Cas9 system for genome editing in Y. lipolytica using a single plasmid (pCAS1yl or pCAS2yl) to transport Cas9 and relevant guide RNA expression cassettes, with or without donor DNA, to target genes. Two Cas9 target genes, TRP1 and PEX10, were repaired by non-homologous end-joining (NHEJ) or homologous recombination, with maximal efficiencies in Y. lipolytica of 85.6 % for the wild-type strain and 94.1 % for the ku70/ku80 double-deficient strain, within 4 days. Simultaneous double and triple multigene editing was achieved with pCAS1yl by NHEJ, with efficiencies of 36.7 or 19.3 %, respectively, and the pCASyl system was successfully expanded to different Y. lipolytica breeding strains. This timesaving method will enable and improve synthetic biology, metabolic engineering and functional genomic studies of Y. lipolytica.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Yarrowia lipolytica is a generally recognized as safe (GRAS) organism and a well-studied unconventional yeast species [24, 29]. It is widely used in multiple industrial fields to produce valuable biochemicals, including organic acids [24] and omega-3 eicosapentaenoic acids (EPA) [44]. The metabolic engineering of Y. lipolytica is heavily documented [13, 22, 26, 38, 44, 45, 47], but traditional genome-editing tools, such as the ura3-blaster [26] and Cre-lox [12] systems, make the process laborious and time-consuming.
Clustered regularly interspaced short palindromic repeats (CRISPR) were originally discovered as an immunological mechanism in bacteria and archaea [2, 42]. The type II CRISPR-Cas9 system has been extensively developed as a powerful genome-editing tool because of its high on-target efficiency in numerous prokaryotes and eukaryotes, including (but not limited to) E. coli [16], Streptomyces spp. [7], Clostridium cellulolyticum [43], Lactobacillus reuteri [30], Saccharomyces cerevisiae [11], Bombyx mori [41], Filamentous fungi [23, 28], Drosophila [46], Candida albicans [40], higher plants [32], and multiple human cell lines [8, 19, 25]. The CRISPR-associated protein (Cas9) endonuclease is guided by a mature CRISPR RNA (crRNA) and a trans-activating crRNA (tracrRNA) towards a target DNA sequence (known as a protospacer). The dual-tracrRNA:crRNA also can be engineered as a single guide RNA (sgRNA) to direct sequence-specific Cas9 dsDNA cleavage [18]. A protospacer-adjacent motif (PAM) immediately follows the 3′ end of the protospacer (NGG in Streptococcus pyogenes, where N represents any nucleotide) [10]. In eukaryotes, DNA double-stranded breaks (DBSs) are induced by the Cas9–RNA complex and are repaired by either homologous recombination (HR) or non-homologous end joining (NHEJ). The NHEJ pathway recruits multiple proteins, including KU70 and KU80, and does not require a homologous template [9]. When a homologous template is present, the HR pathway recruits multiple proteins to repair the DSBs through a homology-based repair process [36]. It has been previously reported that frequency of NHEJ-mediated DSBs repair is high in Y. lipolytica [31].
CRISPR-Cas9 systems have been implemented in three species of yeast, S. cerevisiae [11], Schizosaccharomyces pombe [15] and Kluyveromyces lactis [14]. These tools have enabled single-gene and simultaneous multigene editing [1, 14, 34]. The CRISPR-Cas9 system used in S. cerevisiae was successfully extended to K. lactis [14]. However, this system could not function in Y. lipolytica (data not shown). It is possible that biological impediments exist that would render the S. cerevisiae constructs inapplicable to Y. lipolytica. Schwartz et al. [33] developed a two plasmid-based CRISPR system, in which the sgRNA was expressed by an architectural synthetic RNA polymerase III promoter, and the system successfully achieved single-gene knockouts and knockins in Y. lipolytica.
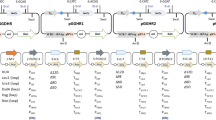
In this study, we demonstrated a single plasmid-based CRISPR-Cas9 system using pCASyl that successfully achieved single or multiple gene disruption via NHEJ or HR in wild-type, ku70-deficient and ku70/ku80 double-deficient strains of Y. lipolytica (Fig. 1). The pCASyl can be easily cured to enable multiple rounds of gene editing. The pCASyl system was successfully expanded to different Y. lipolytica strains for gene disruption.

Overview of genome editing by the CRISPR system in Y. lipolytica. a Cells transformed with pCAS1yl without donor DNA, inducing DSBs that were repaired by NHEJ. b Cells transformed with pCAS2yl with donor DNA, inducing DSBs that were repaired by HR. NHEJ non-homogeneous end-joining, HR homologous recombination, DSB double-stranded break. pCASyl plasmids derived from pMCSCen1 backbone. The LEU2 was used for selection in Y. lipolytica and the AmpR was used in E.coli
Materials and methods
Strains, media and culture conditions
The Y. lipolytica strains used in this study are listed in Table 1. The cells used for transformations were cultivated overnight in culture tubes and transferred to culture flasks for incubation and continuous shaking in a fresh YPD medium, as previously described [13]. After transformation, the cells were grown in an appropriate synthetic complete (SC) medium without the auxotrophic compound supplemented by the plasmids [13]. SC-ura (SC-uracil) plates were used to select the ku70- and ku80-disrupted cells. SC-leu (SC-leucine) plates were used to select the pMCSCen1 plasmid and to derive the transformants. SCG-leu indicates that glucose was the unique carbon source in the SC-leu medium. SCO-leu indicates that oleic acid was the unique carbon source in the SC-leu medium. Glycerol was the unique carbon source in the SCGly-leu medium. All Y. lipolytica cells were cultured at 30 °C. Continuous shaking of the liquid cultures was performed at 250 rpm. To cure the pCASyl plasmid, a single colony was cultured for approximately 15 h up to early log-phase, with cell densities reaching an optical absorbance of approximately 0.5 at 600 nm. The culture was diluted to approximately 1000 cells per milliliter and was plated onto YPD agar.
Construction of plasmids
Plasmids are listed in Table 1; primers are listed in Table S1.
HisG-URA3-hisG cassette construction
(1) The HisG-BamHI-F and HisG-HindIII-R primer set was used to amplify a 1.1-kb hisG fragment from Salmonella genomic DNA. The resulting hisG fragment was digested with BamHI and HindIII and inserted into the pTA vector. (2) The URA3 cassette fragment was amplified from NRRL Y-1095 genomic DNA with the Ura3-KpnI-F and Ura3-BamHI-R primer set and was digested with BamHI and KpnI for insertion into the plasmid generated in step 1. (3) Another hisG fragment was amplified with the HisG-EcoRI-F/HisG-KpnI-R primer set and was digested with KpnI and EcoRI for insertion into the plasmid generated in step 2, yielding the pTA-HUH plasmid.
ku70 and ku80 deletion cassette construction
For ku70, the upstream and downstream homologous arms were amplified from MYA2613 genomic DNA using the KU70-UP-F/R and KU70-DN-F/R primer pairs, respectively. This was followed by overlap extension polymerase chain reaction (OE-PCR) mediated assembly to generate a fusion fragment. The fusion fragment was cloned into pTAs to generate pTAs-ku70. The larger fragment, hisG-ura3-hisG, was excised from pTAs-hisGura3hisG with EcoRI and HindIII and ligated via EcoRI/HindIII into the pTAs-ku70 backbone to generate the pTAs-ku70-HUH plasmid. pTAs-ku70-HUH was digested by SwaI to generate a larger fragment (the cassette for the ku70 knockout), which was used for MYA-2613 transformation.
For ku80, the upstream and downstream homologous fragments were amplified from MYA-2613 genomic DNA with the Ku80-up-F/Ku80-up-R and Ku80-dn-F/Ku80-dn-R primer pairs, respectively. URA3 was amplified using the URA3-△ku80-F/URA3-△ku80-R primer set and was assembled with the upstream and downstream homologous fragments by OE-PCR to generate the Ku80 deletion cassette for the transformation used for the ku80 deletion.
Cas9 expression cassette
(1) Three fragments were amplified from MYA-2613 genomic DNA and a p415-GalL-Cas9-CYC1t template using the pTEFin-Sal1-F/pTEFin-hCas9-OE-R, hCas9-TEFin-OE-F/hCas9-Sal1 mutation-R and hCas9-Sal1 mutation-F/hCas9-region BamH1-Mlu1-R primer pairs. (2) The three fragments were assembled by OE-PCR and cloned into pTAs. A 2336-bp fragment, generated by digesting p415-GalL-Cas9-CYC1t with BamHI and MluI, was inserted into pTAs-TinhC to generate pTAs-TinCas9, which contained the Cas9 expression cassette. The 4875-bp Cas9 expression cassette was generated by digesting pTAs-TinCas9 with SalI and MluI.
CRISPR plasmid construction
The backbone was amplified using the pCen1-Sal1-F/pCen1-Mlu1-R primer set to include the MluI and SalI sites to linearize the backbone, resulting in a 59-bp deletion from 652 to 710 bp. The 4875-bp Cas9 expression cassette (Fig. S1) was ligated into the backbone that was previously digested by SalI and MluI, yielding the plasmid pCen1-Cas9.
sgRNA expression cassette construction
The sgRNA-HDV sequence was ordered from Genscript (Nanjing, Jiangsu, CN). TEFin-HH-sgRNA-HDV-mig1t was generated by assembling the TEFin fragment (amplified with TEF1p-Pme1-F/TEF-TRP1.1-HH-R) with the HH-g RNA-HDV fragment (amplified with TRP1.1-HH-F/HDV-mig1t-R) and the mig1t terminator (amplified with Mig1t-HDV-F/Mig1t-Sal1-R). The assembly fragments, sgRNA expression cassette (Fig. S2), and pCen1-Cas9 backbone were ligated together after being digested with SalI and MssI to generate the pCAS1yl-trp plasmid (Addgene No. 73226). The upstream and downstream regions of the donor DNA were fused with the sgRNA expression cassette fragment by OE-PCR, and the fusion products were ligated with pCen1-Cas9 after digestions with SalI and MssI. The resulting plasmid was pCAS2yl-trp (Fig. S3). The pCASyl-pex plasmids were constructed as described for pCASy-trp.
The TRP1 sgRNA expression cassette fragment, which was amplified using the TEFin-Pme1Sph1-F/Mig1t-Avr2Sal1-R primer set to establish MssI/SphI and AvrII/SalI sites at its 5′ and 3′ ends, respectively, was ligated into pCen1-Cas9 via SalI/MssI to generate the pCAS1y-trp(Sph1Avr2) plasmid (Fig. S4A). The PEX10 sgRNA expression cassette fragment was amplified using the TEFin-Avr2-F/Mig1t-Sal1-R primer set and digested with AvrII and SalI. The AvrII/SalI fragment was ligated into the pCAS1y-trp(Sph1Avr2) vector backbone via AvrII/SalI to generate pCAS1y-TP (Fig. S4B). The GUT2 sgRNA expression cassette was amplified with TEF1p-Pme1-F/Mig1t-Sph1-R, digested with MssI and SphI and inserted into pCAS1y-TP via MssI/SphI to generate pCAS1y-GTP (Fig. S4C). The PEX10 and GUT2 guide sequences used in this study are shown in Fig. S5.
Yeast transformation
The transformation procedure was performed as previously described [13]. Briefly, the 50-μL transformation mixture was plated onto SC-leu solid media for colony counting and for the calculation of transformation efficiency. The remaining transformation mixture was transferred to a tube containing SC-leu liquid media for 2–4 days incubation, and the culture was diluted as needed for plating onto SC-leu solid media.
Phenotype verification
Colonies from the SC-leu plate were randomly picked and streaked onto SC-leu and SC-leu-trp plates, and incubated at 30 °C for 2 days. SCG-leu and SCO-leu media were used in the PEX10 experiments; the SCGly-leu medium was used in the GUT2 experiments.
Colony PCR confirmation and sequencing
The high efficient and success rate DNA polymerase KOD FX Neo (TOYOBO, CO.,LTD) was used for confirmation by colony PCR. The products were sequenced as needed by Sangon Biotech (Shanghai) Co., Ltd. The TRP1-ORF-YZ-F/TRP1-YZ-R primer set was used for confirmation by PCR for the TRP1 experiments. The PEX10-donor-440up-F/PEX10-dn-YZ-R primer set was used for confirmation by PCR for the PEX10 experiments. The Gut2-donor-470up-F/△GUT2-YZ-dn-R primer set was used for gut2 confirmation by colony PCR. All PCR products were sequenced by Sangon Biotech (Shanghai) Co., Ltd.
Results and discussion
Construction of the Y. lipolytica CRISPR system
Because NHEJ-mediated DSB repair dominant Y. lipolytica [31] and can be weakened by disrupting one or both of the ku70 and ku80 genes [20, 39], two CRISPR-Cas9 plasmids were developed with or without donor DNA. pCAS1yl was designed by utilizing a codon-optimized nuclease Cas9 gene from S. pyogenes MGAS5005 [11] that was controlled by a strong, endogenous TEFin promoter [37], a sgRNA containing a 20-bp guide sequence flanked upstream by the hammerhead (HH) ribozyme and downstream by the hepatitis delta virus (HDV) ribozyme [28] (transcribed by the TEFin promoter), and a replicative vector backbone (pMCSCen1) [4] (Fig. 1). pCAS2yl was designed by inserting the donor DNA upstream of the sgRNA expression cassette (Fig. 1). Three different host strains were examined, and they included (1) wild-type MYA-2613 (WT), (2) MYA-2613△ku70 (△ku70) and (3) MYA-2613△ku70△ku80 (△ku70△ku80).
The CRISPR-Cas9 system was initially tested for Cas9-mediated toxicity. The total colony-forming units (cfu) were calculated based on the number of colonies that grew on the SC-leu agar. There were no apparent differences between the transformation efficiencies of the plasmid containing Cas9 and the plasmid containing an inactivated Cas9 gene, indicating that Cas9 had no effect on Y. lipolytica growth (Fig. 2).

CRISPR-Cas9-mediated death of Y. lipolytica cells. Relative transformation efficiency with inactivated Cas9, Cas9, and Cas9 + TRP1.gRNA. Inactive Cas9, the pCen1-inCas9 (11.2 kb) plasmid containing an inactivated cas9 mutant was used as the null control. Cas9 the pCen1-Cas9 (10.7 kb) plasmid carrying a Cas9 expression cassette. Cas9 + trp1.g RNA, the pCAS1yl-trp (11.9 kb) plasmid carrying Cas9 and the sgRNA gene expression cassettes, which targeted to the TRP1. One microgram per plasmid was used for the transformation. The transformation mixture without the outgrowth step was plated on SC-leu agar. The values and error bars represent the average readings and standard deviations for three experiments
The TRP1 gene, which encodes N-(5-phosphoribosyl)-anthranilate isomerase, was selected as a target for editing. When Cas9 was expressed with the guide RNA targeted the TRP1 locus in the genome, the cfu values dramatically decreased in the WT, △ku70 and △ku70△ku80 strains (31.7 ± 5, 17.2 ± 2.3 and 11.4 ± 1.7 %, respectively, Fig. 2). The differences in the survival rates may be due to the NHEJ-mediated repairs of the DSBs generated by Cas9 in the WT and △ku70 strains [9]. The low survival rate observed in the △ku70△ku80 strain was a likely result of insufficient Cas9 or sgRNA activity [1, 11].
Disruption of TRP1 by the pCAS1yl system via NHEJ in the WT and ku70 deficient strains
When donor DNA is unavailable in Y. lipolytica, Cas9-induced DSBs can be repaired only by NHEJ, which involves nonspecific insertions or deletions. This results in target gene mutations and precludes continual cleavage by Cas9 [11, 23, 28, 40].
Inactivation of TRP1 (trp −) resulted in Y. lipolytica tryptophan auxotrophy, and the trp − mutant was unable to grow on a synthetic complete (SC) medium lacking tryptophan [6]. The disruption efficiency achieved with pCAS1yl-trp was calculated based on the number of cells on the agar plate with or without tryptophan. Without outgrowth, a disruption efficiency of 12.5 ± 7.4 % (15/84, 5/69) was observed for the WT strain (Fig. 3a, b), and no mutations were detected in the △ku70 (0/54, 0/40) and △ku70△ku80 (0/26, 0/52) strains (Fig. 3a).

CRISPR-Cas9-mediated TRP1 gene disruption by NHEJ. a TRP1 gene disruption efficiency in WT, △ku70 and △ku70△ku80 via pCAS1yl-trp. b A representative WT transformant phenotype-confirmation is shown. All confirmations were conducted following a similar procedure. Transformants were seeded onto agar with (+trp) or without (−trp) tryptophan. c A representative sample is shown for the alignments of the TRP1 gene sequence from selected trp − mutants in WT Y. lipolytica. Indels were induced, resulting in TRP1 disruptions, and were indicated in red color. The values and error bars represent the average readings and standard deviations for two experiments
A previous study showed that disruption efficiency clearly improved after an extended outgrowth step following transformation [1]. We tested cell outgrowth times of 2 and 4 days before plating for WT, which resulted in efficiencies of 62.5 ± 38.5 % (30/85, 35/39) and 85.6 ± 7.1 % (66/88, 29/32), respectively. In the ku70-deficient strain, the disruption efficiencies were 1.7 ± 0.9 % (1/97, 2/82) for the 2-day outgrowth and 26.1 ± 19.6 % (16/40, 5/41) for the 4-day outgrowth (Fig. 3a). However, no trp − mutants (0/88, 0/34) were detected in the ku70 and ku80 double-deficient strains (Fig. 3a), indicating that KU factor deletions also drastically decreased NHEJ in Y. lipolytica [9]. More than 100 mutants of auxotrophic for tryptophan were sequenced in our work. The sequencing results showed that 100 % of colonies had indels in expected TRP1 gene locus as some were listed in Fig. 3c. When sgRNA was absent, no trp − mutants were detected (Fig. S6).
Deletion of the TRP1 gene via HR in ku70-deficient or ku70/ku80 double-deficient Y. lipolytica
To determine the HR frequency, we initially co-transformed pCAS1yl-trp and linear donor DNA. Without the outgrowth, the HR efficiency was approximately 10 % in the WT trp − mutants, and there was no improvement with 2 days of outgrowth. For Δku70 and Δku70Δku80, the frequencies of the trp − mutation were under 10 % using the approach described above (Fig. 4). We speculated that the linear DNA was not sufficiently sustained inside the cell, thus hampering HR after outgrowth. Therefore, we delivered the donor DNA using a replicative plasmid, as previously described [1, 17]. The donor DNA containing 500 bp upstream and 500 bp downstream sequences of TRP1 was fused with the sgRNA expression cassette and cloned into pCen1-Cas9 to form pCAS2yl-trp (Fig. S3).

CRISPR-Cas9 mediated HR. A Disruption efficiency of the TRP1 gene mediated by CRISPR-Cas9 with different outgrowth times. B Percentage of TRP1 gene disruption via HR in trp − mutants. a pCAS1yl plus the co-transformed linear donor DNA; b pCAS2yl alone transformed. Mean ± standard deviation, n = 2. # These detections were not performed
The phenotypes of the transformants with pCAS2yl-trp were assessed as described above, and their genotypes were confirmed by colony PCR. After 4 days of outgrowth, total trp − mutation efficiencies of 72.3 ± 15 % (54/88, 68/82), 58.9 ± 3.5 % (49/87, 54/88) and 94.1 ± 4.3 % (79/81, 80/88) via NHEJ or HR were observed for WT, △ku70 and △ku70△ku80, respectively (Fig. 4a). The colony PCR results indicated that only 11.1 ± 3.6 % (7/51, 4/47) were generated by HR in the WT trp − mutants, whereas HR-mediated efficiencies were increased to 100 % in the two NHEJ-defective strains (△ku70, 3/3,16/16, △ku70△ku80, 2/2, 13/13) (Figs. 4b, S3B). When sgRNA was absent in these two above conditions, there were no trp − mutants being detected (Figs. S7, S8).
PEX10 deletion with pCASyl and expansion of the pCASyl system to more breeding strains
We investigated whether our CRISPR-Cas9 system could be expanded to other genes and Y. lipolytica strains by focusing on PEX10, another key metabolic gene [5, 33, 44]. PEX10 disruption causes a cell to lose its ability to grow on medium containing oleic acid as the unique carbon source. In this study, WT and △ku70△ku80 were transformed with pCAS1yl-pex and pCAS2yl-pex, respectively, to target the PEX10 gene. pCAS2yl-pex contained the donor DNA, which consisted of the following PEX10 components: a 444-bp upstream sequence, a 450-bp downstream sequences (Fig. S4A) and the intended 200-bp deletion (Fig. S4B). The resulting mutation efficiencies for MYA-2613 and △ku70△ku80 were 62.5 ± 21.4 % (17/22, 16/34) and 28.3 ± 2.4 % (3/10, 4/15), respectively (Table 2; Figs. S9, S10). Additionally, TRP1 was successfully disrupted in the ATCC201249 strain derived from the E122 parent strain [35], unlike the MYA-2613 parent strain (W29) [21], efficiency was 98.1 ± 2.5 % (2/2, 26/27) (Table 2).
One-step multiplex gene disruption with pCAS1yl system
Simultaneous double and triple multigene disruption efficiencies were also investigated for the pCAS1yl system. At first, we tried two plasmids (pCAS1yl-trp and pCAS1yl-pex) co-transformation into the WT strain for TRP1 and PEX10 disruption. However, only 1.6 % (1/60) double gene disruption efficiency was achieved (Fig. S11), while single gene disruption efficiency reached 40 % (TRP1, 24/60) or 56.6 % (PEX10, 34/60). The pCAS1yl-TP plasmid (Fig. S4B), which contained two tandem sgRNA expression cassettes, was thus constructed. A double-disruption efficiency of 36.7 ± 8.5 % (18/42, 12/39) was achieved in 4 days, which was drastically higher than that of the two-plasmid co-transformation procedure (Fig. S12A). The sequencing results indicated that indels were introduced into both target loci (Fig. S12B). To determine whether the system enables simultaneous three targets, an additional GUT2(YALI0B13970g) gene, encoding the mitochondrial sn-glycerol-3-phosphate dehydrogenase, was chosen as the third target gene. The mutant that contained an inactivated gut2 exhibited growth on glucose but not on glycerol [3, 27]. The pCAS1yl-GTP plasmid (Fig. S4C), carrying an sgRNA in tandem targeting GUT2, TRP1 and PEX10 gene cassettes, was constructed and transformed into the WT strain. A triple-gene disruption efficiency of 19.3 ± 9.2 % (5/39, 8/31) was achieved after 4 days, and the mutation was confirmed by sequencing (Fig. 5).

Triple simultaneous disruption. Colonies were picked from transformant plates and diluted in 10 μL of sterilized water were inoculated onto agar plates containing one of four different media to confirm phenotypes. Triple-disruptants only grew on SCG-leu. a Representative image of the phenotype confirmations for the GTP triple disruptants. Green lines indicate positive disruptants. b Alignments of gene sequences from five disruptants shown in a against wild-type sequences. Red indicates variations from the wild-type sequence (WT). One of two replicates is shown
Multi-round genome editing by plasmid curing
To enable multiple rounds of genome editing, plasmid curing was necessary [17]. The pCAS1yl transformant was cultured in non-selective liquid medium (YPD) and plated on YPD agar. Colonies were picked from a plate and confirmed by replica dripping on SC medium agar (+leu) and SC-leu medium agar (−leu). The resulting frequency of colonies that do not grow on -leu from the total was calculated as 38 % (24/63) (Fig. S13A). We observed that smaller colonies on YPD medium did not grow on medium lacking leu (−leu) (Fig. S13B), which agreed with the previous report [12]. The isolations of colonies from +leu and −leu plates were further transformed into E. coli DH5α. We did not get any E. coli transformants through those isolates from colonies which only grew on +leu (Fig S13C), indicating the loss of plasmid pCAS1yl. Thus, single, double or triple target modification could be accomplished via pCASyl within 6 days, and 2 additional days were needed for pCASyl curing to realize next round of genome modification (Fig. S14).
Conclusions
In this report, we have described the development of a pCASyl-based CRISPR-Cas9 system for Y. lipolytica. This genetic tool enabled efficient, scarless, single or multigene editing in several Y. lipolytica strains through NHEJ and HR. The pCASyl CRISPR-Cas9 system established here is more efficient than traditional genome-editing methods and will strongly facilitate synthetic biology, metabolic engineering and functional genomic studies of Y. lipolytica.
References
Bao Z, Xiao H, Liang J, Zhang L, Xiong X, Sun N, Si T, Zhao H (2015) Homology-integrated CRISPR–Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth Biol 4:585–594
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712
Beopoulos A, Mrozova Z, Thevenieau F, Le Dall MT, Hapala I, Papanikolaou S, Chardot T, Nicaud JM (2008) Control of lipid accumulation in the yeast Yarrowia lipolytica. Appl Environ Microbiol 74:7779–7789
Blazeck J, Liu L, Redden H, Alper H (2011) Tuning gene expression in Yarrowia lipolytica by a hybrid promoter approach. Appl Environ Microbiol 77:7905–7914
Blazeck J, Hill A, Liu L, Knight R, Miller J, Pan A, Otoupal P, Alper HS (2014) Harnessing Yarrowia lipolytica lipogenesis to create a platform for lipid and biofuel production. Nat Commun 5
Cheon SA, Han EJ, Kang HA, Ogrydziak DM, Kim J-Y (2003) Isolation and characterization of the TRP1 gene from the yeast Yarrowia lipolytica and multiple gene disruption using a TRP blaster. Yeast 20:677–685
Cobb RE, Wang Y, Zhao H (2015) High-efficiency multiplex genome editing of streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol 4:723–728
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Davis AJ, Chen DJ (2013) DNA double strand break repair via non-homologous end-joining. Transl Cancer Res 2:130–143
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J et al (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607
DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41:4336–4343
Fickers P, Le Dall MT, Gaillardin C, Thonart P, Nicaud JM (2003) New disruption cassettes for rapid gene disruption and marker rescue in the yeast Yarrowia lipolytica. J Microbiol Methods 55:727–737
Gao S, Han L, Zhu L, Ge M, Yang S, Jiang Y, Chen D (2014) One-step integration of multiple genes into the oleaginous yeast Yarrowia lipolytica. Biotechnol Lett 36:2523–2528
Horwitz Andrew A, Walter Jessica M, Schubert Max G, Kung Stephanie H, Hawkins K, Platt Darren M, Hernday Aaron D, Mahatdejkul-Meadows T et al (2015) Efficient multiplexed integration of synergistic alleles and metabolic pathways in yeasts via CRISPR-Cas. Cell Syst 1:88–96
Jacobs JZ, Ciccaglione KM, Tournier V, Zaratiegui M (2014) Implementation of the CRISPR-Cas9 system in fission yeast. Nat Commun 5
Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA (2013) RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239
Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S (2015) Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol 81:2506–2514
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J (2013) RNA-programmed genome editing in human cells. eLife 2: e00471
Kretzschmar A, Otto C, Holz M, Werner S, Hübner L, Barth G (2013) Increased homologous integration frequency in Yarrowia lipolytica strains defective in non-homologous end-joining. Curr Genet 59:63–72
Le Dall M-T, Nicaud J-M, Gaillardin C (1994) Multiple-copy integration in the yeast Yarrowia lipolytica. Curr Genet 26:38–44
Liu G-L, Li Y, Zhou H-X, Chi Z-M, Madzak C (2012) Over-expression of a bacterial chitosanase gene in Yarrowia lipolytica and chitosan hydrolysis by the recombinant chitosanase. J Mol Catal B Enzym 83:100–107
Liu R, Chen L, Jiang Y, Zhou Z, Zou G (2015) Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov 1:15007
Coelho MAZ, Amaral PFF, Belo I (2010) Yarrowia lipolytica an industrial workhorse
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Matthäus F, Ketelhot M, Gatter M, Barth G (2013) Production of lycopene in the non-carotenoid producing yeast Yarrowia lipolytica. Appl Environ Microbiol. doi:10.1128/aem.03167-13
Mori K, Iwama R, Kobayashi S, Horiuchi H, Fukuda R, Ohta A (2013) Transcriptional repression by glycerol of genes involved in the assimilation of n-alkanes and fatty acids in yeast Yarrowia lipolytica. FEMS Yeast Res 13:233–240
Nødvig CS, Nielsen JB, Kogle ME, Mortensen UH (2015) A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS One 10:e0133085
Nicaud J-M (2012) Yarrowia lipolytica. Yeast 29:409–418
Oh J-H, van Pijkeren J-P (2014) CRISPR–Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res 42:e131
Richard G-F, Kerrest A, Lafontaine I, Dujon B (2005) Comparative genomics of hemiascomycete yeasts: genes involved in dna replication, repair, and recombination. Mol Biol Evol 22:1011–1023
Schaeffer SM, Nakata PA (2015) CRISPR/Cas9-mediated genome editing and gene replacement in plants: transitioning from lab to field. Plant Sci 240:130–142
Schwartz CM, Hussain MS, Blenner M, Wheeldon I (2015) Synthetic RNA polymerase III promoters facilitate high efficiency CRISPR-Cas9 mediated genome editing in Yarrowia lipolytica. ACS Synth Biol. doi:10.1021/acssynbio.5b00162
Shi S, Liang Y, Zhang MM, Ang EL, Zhao H (2016) A highly efficient single-step, markerless strategy for multi-copy chromosomal integration of large biochemical pathways in Saccharomyces cerevisiae. Metab Eng 33:19–27
Smith JJ, Szilard RK, Marelli M, Rachubinski RA (1997) The peroxin Pex17p of the yeast Yarrowia lipolytica is associated peripherally with the peroxisomal membrane and is required for the import of a subset of matrix proteins. Mol Cell Biol 17:2511–2520
Sung P, Klein H (2006) Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 7:739–750
Tai M, Stephanopoulos G (2013) Engineering the push and pull of lipid biosynthesis in oleaginous yeast Yarrowia lipolytica for biofuel production. Metab Eng 15:1–9
Theerachat M, Emond S, Cambon E, Bordes F, Marty A, Nicaud J-M, Chulalaksananukul W, Guieysse D et al (2012) Engineering and production of laccase from Trametes versicolor in the yeast Yarrowia lipolytica. Bioresour Technol 125:267–274
Verbeke J, Beopoulos A, Nicaud J-M (2013) Efficient homologous recombination with short length flanking fragments in Ku70 deficient Yarrowia lipolytica strains. Biotechnol Lett 35:571–576
Vyas VK, Barrasa MI, Fink GR (2015) A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv 1
Wang Y, Li Z, Xu J, Zeng B, Ling L, You L, Chen Y, Huang Y et al (2013) The CRISPR/Cas system mediates efficient genome engineering in Bombyx mori. Cell Res 23:1414–1416
Wiedenheft B, Sternberg SH, Doudna JA (2012) RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338
Xu T, Li Y, Shi Z, Hemme CL, Li Y, Zhu Y, Van Nostrand JD, He Z et al (2015) Efficient genome editing in Clostridium cellulolyticum via CRISPR-Cas9 Nickase. Appl Environ Microbiol 81:4423–4431
Xue Z, Sharpe PL, Hong S-P, Yadav NS, Xie D, Short DR, Damude HG, Rupert RA et al (2013) Production of omega-3 eicosapentaenoic acid by metabolic engineering of Yarrowia lipolytica. Nat Biotech 31:734–740
Ye R, Sharpe P, Zhu Q (2012) Bioengineering of oleaginous yeast Yarrowia lipolytica for lycopene production. In: Barredo JL (ed) Microbial carotenoids from fungi. Humana Press, New York, pp 153–159
Yu Z, Ren M, Wang Z, Zhang B, Rong YS, Jiao R, Gao G (2013) Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics. doi:10.1534/genetics.113.153825
Zhu Q, Jackson EN (2015) Metabolic engineering of Yarrowia lipolytica for industrial applications. Curr Opin Biotechnol 36:65–72
Acknowledgments
We would like to thank Professor Hal S. Alper for the generous gift of plasmid pMCSCen1. This study was financed by the Ministry of Science and Technology of China (973:2012CB721105; 863:2012AA02A704). This study was also supported in part by project (973:2014CB745101) and the STS project from CAS (KFJ-EW-STS-030).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gao, S., Tong, Y., Wen, Z. et al. Multiplex gene editing of the Yarrowia lipolytica genome using the CRISPR-Cas9 system. J Ind Microbiol Biotechnol 43, 1085–1093 (2016). https://doi.org/10.1007/s10295-016-1789-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-016-1789-8