
Abstract
Bone morphogenetic protein 7 (BMP-7) is a multifunctional differentiation factor that belongs to the transforming growth factor superfamily. BMP-7 induces gene expression of protein tyrosine phosphatase-like, member A/cementum attachment protein (PTPLA/CAP) and cementum protein 1 (CEMP1), both of which are markers of cementoblasts and cementocytes. In the previous study, we reported that BMP-7 treatment enhanced PTPLA/CAP and CEMP1 expression in both normal and immortal human periodontal ligament (PDL) cells. To elucidate the molecular mechanisms of the gene expression of these molecules, in this study, we identified a functional transcription activator binding region in the promoter region of PTPLA/CAP and CEMP1 that is responsive to BMP signals. Here, we report that some short motifs termed GC-rich Smad-binding elements (GC-SBEs) that are located in the human PTPLA/CAP promoter and CEMP1 promoter are BMP-7 responsive as analyzed with luciferase promoter assays. On the other hand, we found that transcription of Sp7/Osterix and PTPLA/CAP was up-regulated after 1 week of BMP-7 treatment on purified normal human PDL cells as a result of gene expression microarray analysis. Furthermore, transcription of Sp7/Osterix, runt-related transcription factor 2 (RUNX2), and alkaline phosphatase (ALP) was up-regulated after 2 weeks of BMP-7 treatment, whereas gene expression of osteo/odontogenic markers such as integrin-binding sialoprotein (IBSP), collagen, type I, alpha 1 (COL1A1), dentin matrix acidic phosphoprotein 1 (DMP1), and dentin sialophosphoprotein (DSPP) was not up-regulated in purified normal or immortal human PDL cells as a result of qRT-PCR. The results suggest that BMP-7 mediates cementogenesis via GC-SBEs in human PDL cells and that its molecular mechanism is different from that for osteo/odontogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cementum is the mineralized connective tissue that covers the root surface of the tooth and provides the interface, through which the tooth root surface is anchored to the Sharpey’s fibers [1, 2]. Cementogenesis is initiated after dentin formation of the tooth root and is regulated by the interaction between Hertwig’s epithelial root sheath and dental follicle-derived mesenchymal cells [3]. PDL cells are mesenchymal cells that originate from the dental follicle that forms during the cap stage of tooth germ development [4]. These dental follicle-derived cells are thought to contain undifferentiated, lineage-committed cells in their population [5]. Previous studies have reported that dental follicle-derived mesenchymal stem/progenitor cells acquire cementoblast functions following stimulation with signaling molecules including bone morphogenetic protein (BMP)-2 and -7, which activate the BMP pathway [6–9].
BMPs, which belong to the transforming growth factor (TGF) superfamily, are multifunctional differentiation factors [10]. BMP-7, which is also known as osteogenic protein 1, is a multifunctional cytokine. During tooth root morphogenesis, BMP-7 is localized in alveolar bone, cementum, and the PDL, whereas BMP-2 is localized only in the alveolar bone [11]. During cementoblast differentiation, BMP-2 inhibits differentiation and mineralization in culture [12]. BMP-7 induces cementogenesis [13]. However, the molecular mechanisms of cementoblast differentiation and mineralization through the BMP pathway remain largely unclear.
We previously reported that a small population of cells purified from human PDL cells, which are positive for putative surface antigens of mesenchymal stem cells, strongly express protein tyrosine phosphatase-like, member A/cementum attachment protein (PTPLA/CAP) and cementum protein 1 (CEMP1), the latter of which is also known as cementum protein-23 [2]. Both of PTPLA/CAP and CEMP1 are markers of cementoblasts and cementocytes, which are induced by stimulation with BMP-7 [14].
Recently, Smads, that are regulators of TGF-β signal transduction, were reported to bind motifs termed GC-rich Smad-binding elements (GC-SBEs) [15], which are associated with target gene up-regulation in mammals. Several sequences in the identified GC-SBE motifs show relatively weak affinity for Smad binding [15].
In this study, we identified GC-SBE motifs in the promoter region of PTPLA/CAP and CEMP1 and examined their cellular response to BMP-7 stimulation in cultured human PDL cells. When stimulated with BMP-7, these human PDL-derived cells showed high levels of expression for the above markers of cementogenesis.
Materials and methods
Cell culture
As indicated in the previous study [16], we cultured the Pel cells obtained by the orthodontical reason from normal adult human PDL. Then, this crude population of Pel cells was immortalized by transfection with H-TERT gene at 9 passages and was named Pelt cells [17]. These cells could contain a small population of dental follicle-derived somatic stem cells those have cementogenic and osteogenic potential. Those cells were sorted with the FACSAria™ III Flow Cytometry System (BD Biosciences, Franklin Lakes, NJ, USA) using anti-CD44, anti-CD105 (eBioscience, San Diego, CA, USA), and anti-stage-specific embryonic antigen 3 (SSEA-3) antibodies (BioLegend, San Diego, CA, USA) [14]. This procedure resulted in isolation of mesenchymal stem cells from the total population of PDL cells [2]. The sorted cells were then cultured in STEMPRO® MSC SFM (GIBCO/Life Technologies, Carlsbad, CA, USA), which is a medium for mesenchymal stem cell culture. Cells were maintained in a humidified atmosphere of 5 % CO2/95 % air at 37 °C. The medium was changed every 2 days. When the cells reached confluence, they were subcultured at a split ratio of 1:2–1:4 by gentle separation with accutase solution (Innovative Cell Technologies, San Diego, CA, USA) for 4 min at room temperature. The number of population doublings of Pel cells used in these experiments was between 16 and 19. For induction of cementoblastic differentiation, these human PDL-derived cells were cultured in 25-cm2 flasks in STEMPRO® MSC SFM until they reached subconfluence. The medium was replaced with MEM supplemented with 2 % FBS containing 200 ng/ml rhBMP-7, and the cells were cultured for 1 or 2 weeks. Pel cells and Pelt cells cultured without rhBMP-7 served as controls.
DNA constructs
For all plasmid constructs, standard recombinant DNA technologies were used [18]. These reporter vectors Tripluc Vectors (TOYOBO, Osaka, Japan) for luciferase assay contain β-lactamase genes so that the subcloned fragments are identified by selection for ampicillin resistance. To generate the reporter construct pCEMP1107, the 5′–3′ XhoI–HindIII fragment between nucleotides (nt) −1107 and +50 of the human CEMP1 promoter was subcloned into the 5′–3′ XhoI and HindIII sites of the pStable Luciferase Orange (SLO) test vector (TOYOBO, Osaka, Japan). The Herpes simplex virus thymidine kinase (HSVtk) promoter motif from the pStable Luciferase Green (SLG)-HSVtk control vector (TOYOBO) was inserted between the CEMP1 promoter and pSLO to drive stable expression of the reporter gene. The pSLO vector has a variant of the firefly luciferase gene as the downstream reporter, whereas the pSLG vector has the wild-type firefly luciferase gene.
The CEMP1 promoter deletion construct pCEMP391 was generated with a fragment between nt −391 and +50 that was amplified with PCR using specific primers containing XhoI and HindIII sites. The HSVtk promoter motif was also ligated into the 5′–3′ SpeI and EcoRV sites. The pStable Luciferase Red (pSLR) vector has the railroad worm luciferase gene as the downstream reporter. pCAP869 was similarly generated from a fragment between nt −869 and −21 of the human PTPLA/CAP promoter containing the 5′–3′ XhoI and BglII sites. It was subcloned into the 5′–3′ XhoI and BglII sites of the SLR test vector (TOYOBO), and the HSVtk promoter motif was ligated into the 5′–3′ SpeI and EcoRV sites.
The PTPLA/CAP promoter deletion construct pCAP398 was generated from a fragment between nt −869 and −398 containing XhoI and BglII sites. The HSVtk promoter motif was ligated into the 5′–3′ SpeI and EcoRV sites. All constructs were confirmed with DNA sequencing.
Transient transfection
Pelt cells were seeded at a density of 15,000 cells/well in a 96-well white plate (Greiner Bio-One, Frickenhausen, Germany), and after overnight culture, the cells were transfected using Opti-MEM® I Reduced-Serum Medium (GIBCO/Life Technologies) and Lipofectamine® LTX (Life Technologies) transfection reagent in accordance with the manufacturer’s instructions. Transfections were performed with 40 ng pCEMP1107 or pCAP869 and 20 ng pSLG-HSVtk control vector per well. The deletion constructs pCEMP391 and pCAP398 were similarly co-transfected with 20 ng pSLG-HSVtk control vector per well. The pSLG-HSVtk control vector was co-transfected as an internal control to normalize variations in transfection efficiency by dividing the measurement for SLO or SLR activity [19]. All DNAs were prepared using NucleoBond® Xtra Midi EF according to the manufacturer’s instructions (Macherey-Nagel, Düren, Germany).
Luciferase assay
Twenty-four hours post-transfection, the medium was replaced with minimum essential medium (MEM) (GIBCO/Life Technologies) supplemented with 2 % fetal bovine serum (FBS) containing 200 ng/ml recombinant human BMP-7 (rhBMP-7) (R&D Systems, Minneapolis, MN, USA), and the cells were cultured for 12 or 24 h. Luciferase activity was measured using Tripluc® Luciferase Assay Reagent (TOYOBO) with a PHELIOS Luminometer for 96/384-well plates (ATTO, Tokyo, Japan). The results presented are representative data from at least four separate experiments performed in quadruplicate.
Gene expression microarrays
Total RNA from purified SSEA-3-positive Pel cells cultured with or without rhBMP-7 for 1 week was isolated with the RNeasy® Mini Kit according to the manufacturer’s instructions (QIAGEN, Hilden, Germany). The RNA was quantified and quality tested with spectrophotometry and gel electrophoresis. All samples had 260/280 absorbance ratios between 1.8 and 2.1 and showed prominent 18 and 28 s bands. The cRNA was amplified and labeled using a Low Input Quick Amp Labeling Kit (Agilent Technologies, Santa Clara, CA, USA) and hybridized using the SurePrint G3 Human Gene Expression Microarray 8 × 60 K v2 Kit (Agilent Technologies). All hybridized microarray slides were scanned with an Agilent scanner. Relative hybridization intensities and background hybridization values were calculated using Agilent Feature Extraction Software (9.5.1.1).
Data analyses and filter criteria
Raw signal intensities and flags for each probe were calculated from hybridization intensities (gProcessedSignal) and spot information (glsSaturated, etc.) according to the procedures recommended by Agilent. Flag criteria on GeneSpring Software were: Absent (A) “Feature is not positive and significant” and “Feature is not above background”. Marginal (M) “Feature is not Uniform”, “Feature is Saturated”, and “Feature is a population outlier”. Present (P) others. The raw signal intensities of samples (control versus rhBMP-7-treated group) were log2-transformed and normalized with the quantile algorithm using the ‘preprocessCore’ library package [20] in the Bioconductor software [21]. We selected probes that registered the ‘P’ flag in both samples. To identify up- or down-regulated genes, we calculated the intensity-based Z scores [22] and ratios (non-log-scaled fold-change) from the normalized signal intensities of each probe for comparison between the control and experiment sample.
Then we established criteria for changed genes: up-regulated genes: Z score ≥2.0 and ratio ≥1.5-fold; down-regulated genes: Z score ≤−2.0 and ratio ≤0.66. To determine significantly over-represented gene ontology categories and significant enrichment of pathways, we used tools and data provided with Ingenuity Pathway Analysis.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA (2 μg) was isolated and purified from Pel cells and Pelt cells cultured with or without rhBMP-7 for 2 weeks and then used for RT using the High Capacity RNA-to-cDNA Kit (Applied Biosystems/Life Technologies). Reactions were performed in a GeneAmp 2400 thermal cycler (PerkinElmer Life Sciences, Waltham, MA, USA). For qRT-PCR, TaqMan® gene expression assays and the StepOnePlus™ Real-Time PCR System (Applied Biosystems/Life Technologies) were used to amplify the human target genes PTPLA/CAP (assay ID: Hs00171965_m1, gene bank number: NM_014241.3, amplicon length: 74 bp), CEMP1 (assay ID: Hs04185363_s1, gene bank number: NM_001048212.3, amplicon length: 72 bp), alkaline phosphatase (ALP) (assay ID: Hs01029144_m1, gene bank number: NM_000478.4, amplicon length: 79 bp), integrin-binding sialoprotein (IBSP) (assay ID: Hs00173720_m1, gene bank number: NM_004967.3, amplicon length: 95 bp), collagen, type I, alpha 1 (COL1A1) (assay ID: Hs00164004_m1, gene bank number: NM_000088.3, amplicon length: 66 bp), dentin matrix acidic phosphoprotein 1 (DMP1) (assay ID: Hs00189368_m1, gene bank number: NM_004407.3, amplicon length: 88 bp), dentin sialophosphoprotein (DSPP) (assay ID: Hs00171962_m1, gene bank number: NM_014208.3, amplicon length: 67 bp), runt-related transcription factor 2 (RUNX2) (assay ID: Hs00231692_m1, gene bank number: NM_001015051.3, amplicon length: 116 bp), and Sp7/Osterix (assay ID: Hs01866874_s1, gene bank number: NM_152860.1, amplicon length: 104 bp) (Applied Biosystems/Life Technologies). mRNA expression of β-actin (4326315E − 0710012; Applied Biosystems/Life Technologies) served as an internal control for normalization. Relative standard curves were analyzed with StepOne Software v2.1 (Applied Biosystems/Life Technologies). The experiments were performed in quadruplicate.
Statistical analysis
The statistical significance of the difference in gene expression between the control and rhBMP-7-treated groups amplified with qRT-PCR was determined with the Student’s t test or Fisher’s exact test, assuming double-sided independent variance and with p < 0.05 considered significant.
Results
Identification of BMP-7 response elements in the human CEMP1 and PTPLA/CAP promoters
The DNA sequences of the human CEMP1 and PTPLA/CAP promoters were obtained from the human genome database (GenBank™ number AY584596.1 and AY455942.1, respectively). Computer analysis of the nucleotide sequences suggested the presence of SBEs [17], including GGCGCC (SBE1), GGAGCC (SBE2), and GGTGCC (SBE3), which have different enhancer activities within the proximal promoter (Fig. 1a). These promoter regions did not have potential binding sites for activator protein 1, cyclic AMP response element-binding protein, nuclear factor kappa B, or serum response factor (data not shown).

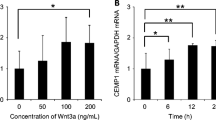
Transient transfection of vectors containing promoters that drive luciferase expression and BMP-7 stimulation of purified Pelt cells. a Human CEMP1 and CAP proximal promoter nucleotide sequences are shown. Several varieties of Smad-binding elements (SBEs) are present in these sequences. b The reporter constructs for triple luciferase assays are shown. c The activities of CEMP1 and CAP promoters that contain SBEs were stimulated with BMP-7. The SBE (−) groups indicate the cells which were transfected the deletion constructs pCEMP391 and pCAP398. The value obtained from the control group no BMP-7 was taken as onefold, and fold increases were calculated by dividing the individual value by the control group value. The graph shows the mean ± SE from four independent experiments performed in quadruplicate. *p < 0.05, BMP-7-treated group compared to BMP-7-untreated group
To assess whether the effects of BMP-7 on CEMP1 and PTPLA/CAP expression were mediated by the SBEs in these promoter regions, we prepared various promoter construct fragments that were ligated to a luciferase reporter gene (Fig. 1b). Pelt cells transfected with the reporter construct pCEMP1107 showed an 8.5-fold increase in transcriptional activity after 12 h of treatment with rhBMP-7 compared with transfectants not treated with rhBMP-7 (Fig. 1c). BMP-7 also induced a 3.0-fold increase in promoter activity in cells transfected with pCAP869. However, cells transfected with the deletion constructs pCEMP391 and pCAP398 did not show significant changes in responsiveness to BMP-7 stimulation (Fig. 1c).
DNA microarray analysis
As shown in Table 1, after 1 week of stimulation with BMP-7, the expression of Sp7/Osterix was 53.25 times higher and PTPLA/CAP was 3.17 higher in BMP-7-treated cells than in control cells. Conversely, purified Pel cells stimulated with BMP-7 did not show significant changes in CEMP1, ALP, COL1A1, or RUNX2 expression. Transcripts for IBSP, DMP1, and DSPP were not detected in any samples (data not shown).
Gene expression related to cementogenesis
As shown in Fig. 2a, after 1 week of stimulation with BMP-7, the expression of CEMP1 was 0.25 times lower in Pelt cells than in control cells. On the other hand, the expression of PTPLA/CAP was 4.21 times higher and 3.98 times higher in Pel and Pelt cells, respectively, than in control cells. In Pel cells, the expression of COL1A1 and ALP did not change significantly after 1 week of BMP-7 treatment. In contrast, in Pelt cells, the expression of COL1A1 and ALP was 4.15 and 9.98 times higher, respectively, than in control cells after 1 week of stimulation with BMP-7. The expression of RUNX2 was 3.75 times higher in Pelt cells stimulated with BMP-7 than in control cells, whereas Pel cells stimulated with BMP-7 did not show significant changes in RUNX2 expression. Both Pel cells and Pelt cells showed more than 60 times higher Sp7/Osterix expression after 1 week of stimulation with BMP-7 compared to control cells. Neither cell line showed significant changes in IBSP expression following stimulation with BMP-7 (data not shown).


Mineralization-related gene expression in Pel cells and Pelt cells. Quantitative RT–PCR analysis was used to determine the gene expression in both cell lines after 1 week (a) and 2 weeks (b) of culture with (rhBMP-7) or without rhBMP-7 (Ctrl). β-actin was used as an internal control. Statistical significance was determined using the Student’s t test (*p < 0.05 vs. control)
As shown in Fig. 2b, after 2 weeks of BMP-7 treatment, the expression of CEMP1 was 1.68 times higher in Pel cells and 2.62 times higher in Pelt cells than in control cells. The expression of PTPLA/CAP was 0.08 times lower in Pel cells stimulated with BMP-7 than in control cells, whereas Pelt cells stimulated with BMP-7 did not show significant changes in PTPLA/CAP expression. Pel cells and Pelt cells stimulated with BMP-7 showed 2.62- and 1.98-fold higher ALP expression, respectively, than control cells. Furthermore, Pel cells and Pelt cells showed 2.42- and 4.11-fold higher RUNX2 expression and 5.36- and 10.48-fold higher Sp7/Osterix expression, respectively, after 2 weeks of stimulation with BMP-7 than control cells. On the other hand, Pel cells and Pelt cells stimulated with BMP-7 did not show significant changes in the expression of COL1A1 (Fig. 2b) or IBSP (data not shown). Transcripts for DMP1 and DSPP were not detected in any samples (data not shown).
Discussion
Conventional surgical periodontal procedures such as guided tissue regeneration techniques and bone grafting have been conducted according to the degree of tissue deficit and lead to reconstruction of periodontal tissue containing alveolar bone, connective tissue, and PDL, which are lost during degenerative changes. Recently, a bioengineered tooth unit was shown to be engrafted and integrated via recipient bone remodeling after transplantation into an extensive bone defect [23]. However, methods for reconstruction of cementum from PDL-derived stem cells have not been established. Furthermore, the molecular mechanisms related to cementogenesis remain unclear. In our present study, we demonstrated that BMP-7 induces cementogenic differentiation of human PDL-derived stem cells via the BMP pathway.
In a recent study, a small population of SSEA-3/CD105 double-positive adult somatic stem cells (1.9 %), somewhat similar to induced pluripotent stem cells, was found among adult human fibroblasts [24]. Another report suggested that human PDL cells also contain a somatic stem cell population with multilineage differentiation potential and showed that SSEA-positive human PDL-derived clonal cells have adipogenic, osteogenic, and chondrogenic potential [25]. More recently, we reported isolation of SSEA-3/CD44/CD105 triple-positive cells from normal and immortal human PDL cells that show cementogenic potential [14]. Therefore, some SSEAs would be specific markers for identification of stem cells in the PDL. During cementogenesis, differentiation of stem/progenitor cells in the dental follicle appears to be regulated by epithelial–mesenchymal interactions that generate specific signals for cellular differentiation [26]. In an in vivo differentiation assay, bovine dental follicle-derived cells were shown to form cementum matrix when transplanted into immunodeficient mice [27]. Murine dental follicle-derived cells were also shown to differentiate along a cementoblast/osteoblast pathway as measured by increased expression of RUNX2/core-binding factor subunit α1 and bone sialoprotein (BSP)/IBSP when stimulated with BMP-2 in vitro [6, 28]. BMP-2 was also reported to induce the expression of PTPLA/CAP in human periodontal ligament cells [29]. Similarly, another study showed that expressions of PTPLA/CAP and cementum protein-23/CEMP1 were detected in cultured human dental follicle cells stimulated with BMP-2 and BMP-7 [7]. We previously reported that human PDL-derived cells and their immortal derivatives stimulated with BMP-7 showed higher levels of expressions of PTPLA/CAP and CEMP1 than the cells stimulated with BMP-2 [14]. However, the details of the stem/progenitor cell biology and the differentiation potential of these cells remain largely unknown.
In osteo/odontogenesis, the progenitors express ALP, BSP, DMP1, and RUNX2, whereas transcription of DSPP is specifically up-regulated during odontogenesis [30–33]. During cementogenesis, the expression of ALP, IBSP, CEMP, PTPLA/CAP, RUNX2, and Sp7/Osterix is up-regulated. However, RUNX2 and Sp7/Osterix transcripts are down-regulated during differentiation of cementoblasts [34–36]. Thus, our findings suggest that isolated PDL-derived cells from Pel cells and Pelt cells show cementoblast differentiation when stimulated with BMP-7, but not terminal differentiation (Fig. 2b). In isolated Pel cells and Pelt cells, the expression of PTPLA/CAP was up-regulated after 1 week of BMP-7 treatment, but not after 2 weeks. Conversely, the expression of CEMP1 was not up-regulated after 1 week of BMP-7 treatment, but it was up-regulated after 2 weeks (Fig. 2a, b). The results suggest that expression of these genes showed a time-delayed response during BMP-7-induced cementogenesis. As a correlate result, we have recently showed the immunocytochemical evidence for the cementoblastic differentiation of purified Pel cells and Pelt cells with rhBMP-7 treatment [14].
The relationship between cementogenesis-related gene expression and BMP signal transduction is not clear. Previous studies have reported that BMP signaling pathways mediate expression of BSP and DSPP via CCAAT-binding factor and regulate osteo/odontogenesis [37, 38]. In the present study, we identified short enhancer sequences termed GC-SBEs [15] that were located in the human PTPLA/CAP and CEMP1 promoters and that were responsive to BMP-7 as analyzed with triple luciferase promoter assays (Fig. 1a–c). Therefore, the present study provides new evidence that BMP-7 activates CEMP1 and PTPLA/CAP transcription via GC-SBEs, and the BMP-7-Smad pathway plays an important role in the mechanism of cementoblast differentiation. Our results provide insight into the molecular mechanism of cementogenesis and may be useful for biological applications using PDL-derived mesenchymal stem cells.
References
Bosshardt DD, Selvig KA. Dental cementum: the dynamic tissue covering of the root. Periodontology. 1997;13:41–75.
Alvarez-Pérez MA, Narayanan S, Zeichner-David M, Rodríguez CB, Arzate H. Molecular cloning, expression and immunolocalization of a novel human cementum-derived protein (CP-23). Bone. 2006;38:409–19.
Bosshardt DD, Schroeder HE. Cementogenesis reviewed: a comparison between human premolars and rodent molars. Anat Rec. 1996;245:267–92.
Chai Y, Jiang X, Ito Y, Bringas P Jr, Han J, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127:1671–9.
Morsczeck C, Gotz W, Schierholz J, Zeilhofer F, Kuhn U, Mohl C, Sippel C, Hoffmann KH. Isolation of precursor cells (PCs) from human dental follicle of wisdom teeth. Matrix Biol. 2005;24:155–65.
Zhao M, Xiao G, Berry JE, Franceschi RT, Reddi A, Somerman MJ. Bone morphogenetic protein 2 induces dental follicle cells to differentiate toward a cementoblast/osteoblast phenotype. J Bone Miner Res. 2002;17:1441–51.
Kémoun P, Laurencin-Dalicieux S, Rue J, Farges JC, Gennero I, Conte-Auriol F, Briand-Mesange F, Gadelorge M, Arzate H, Narayanan AS, Brunel G, Salles JP. Human dental follicle cells acquire cementoblast features under stimulation by BMP-2/-7 and enamel matrix derivatives (EMD) in vitro. Cell Tissue Res. 2007;329:283–94.
Hakki SS, Foster BL, Nagatomo KJ, Bozkurt SB, Hakki EE, Somerman MJ, Nohutcu RM. Bone morphogenetic protein-7 enhances cementoblast function in vitro. J Periodontol. 2010;81:1663–74.
Kitagawa M, Ao M, Miyauchi M, Abiko Y, Takata T. F-spondin regulates the differentiation of human cementoblast-like (HCEM) cells via BMP7 expression. Biochem Biophys Res Commun. 2012;418:229–33.
Ripamonti U, Petit JC. Bone morphogenetic proteins, cementogenesis, myoblastic stem cells and the induction of periodontal tissue regeneration. Cytokine Growth Factor Rev. 2009;20:489–99.
Thomadakis G, Ramoshebi LN, Crooks J, Rueger DC, Ripamonti U. Immunolocalization of bone morphogenetic protein-2 and -3 and osteogenic protein-1 during murine tooth root morphogenesis and in other craniofacial structures. Eur J Oral Sci. 1999;107:368–77.
Zhao M, Berry JE, Somerman M. Bone morphogenetic protein-2 inhibits differentiation and mineralization of cementoblasts in vitro. J Dent Res. 2003;82:23–7.
Jin QM, Zhao M, Economides AN, Somerman MJ, Giannobile WV. Noggin gene delivery inhibits cementoblast-induced mineralization. Connect Tissue Res. 2004;45(1):50–9.
Torii D, Konishi K, Watanabe N, Goto S, Tsutsui T. Cementogenic potential of multipotential mesenchymal stem cells purified from the human periodontal ligament. Odontology. 2014;. doi:10.1007/s10266-013-0145-y (Epub ahead of print).
Morikawa M, Koinuma D, Tsutsumi S, Vasilaki E, Kanki Y, Heldin CH, Aburatani H, Miyazono K. ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res. 2011;39:8712–27.
Omori N, Kobayashi H, Tsutsui T. Quantitative comparison of cytocidal effects of tetracyclines and fluoroquinolones on human periodontal ligament fibroblasts. J Periodont Res. 1999;34:290–5.
Tsutsui T, Kumakura S, Yamamoto A, Kanai H, Tamura Y, Kato T, Anpo M, Tahara H, Barrett JC. Association of p16INK4a and pRb inactivation with immortalization of human cells. Carcinogenesis. 2002;23:2111–7.
Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3rd Ed. Cold Spring Harbor: Cold Spring Harbor Laboratory; 2001. pp. 8.15–8.54.
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, Ohmiya Y. Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques. 2005;38:891–4.
Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80.1–80.16.
Quackenbush J. Microarray data normalization and transformation. Nat Genet. 2002;32(suppl):496–501.
Oshima M, Mizuno M, Imamura A, Ogawa M, Yasukawa M, Yamazaki H, Morita R, Ikeda E, Nakao K, Takano-Yamamoto T, Kasugai S, Saito M, Tsuji T. Functional tooth regeneration using a bioengineered tooth unit as a mature organ replacement regenerative therapy. PLoS One. 2011;6:e21531.
Kuroda Y, Kitada M, Wakao S, Nishikawa K, Tanimura Y, Makinoshima H, Goda M, Akashi H, Inutsuka A, Niwa A, Shigemoto T, Nabeshima Y, Nakahata T, Nabeshima Y, Fujiyoshi Y, Dezawa M. Unique multipotent cells in adult human mesenchymal cell populations. Proc Natl Acad Sci USA. 2010;107:8639–43.
Fukushima H, Kawanabe N, Murata S, Ishihara Y, Yanagita T, Balam TA, Yamashiro T. SSEA-4 is a marker of human deciduous periodontal ligament stem cells. J Dent Res. 2012;91:955–60.
Sharpe PT. Neural crest and tooth morphogenesis. Adv Dent Res. 2001;15:4–7.
Handa K, Saito M, Yamauchi M, Kiyono T, Sato S, Teranaka T, Sampath Narayanan A. Cementum matrix formation in vivo by cultured dental follicle cells. Bone. 2002;31:606–11.
Silvério KG, Davidson KC, James RG, Adams AM, Foster BL, Nociti FH Jr, Somerman MJ, Moon RT. Wnt/β-catenin pathway regulates bone morphogenetic protein (BMP2)-mediated differentiation of dental follicle cells. J Periodont Res. 2012;47:309–19.
Pitaru S, Pritzki A, Bar-Kana I, Grosskopf A, Savion N, Narayanan AS. Bone morphogenetic protein 2 induces the expression of cementum attachment protein in human periodontal ligament clones. Connect Tissue Res. 2002;43:257–64.
Yamazaki H, Tsuneto M, Yoshino M, Yamamura K, Hayashi S. Potential of dental mesenchymal cells in developing teeth. Stem Cells. 2007;25:78–87.
Balic A, Aguila HL, Caimano MJ, Francone VP, Mina M. Characterization of stem and progenitor cells in the dental pulp of erupted and unerupted murine molars. Bone. 2010;46:1639–51.
Simon S, Smith AJ, Lumley PJ, Berdal A, Smith G, Finney S, Cooper PR. Molecular characterization of young and mature odontoblasts. Bone. 2009;45:693–703.
Hao J, Narayanan K, Ramachandran A, He G, Almushayt A, Evans C, George A. Odontoblast cells immortalized by telomerase produce mineralized dentin-like tissue both in vitro and in vivo. J Biol Chem. 2002;277:19976–81.
Saito M, Handa K, Kiyono T, Hattori S, Yokoi T, Tsubakimoto T, Harada H, Noguchi T, Toyoda M, Sato S, Teranaka T. Immortalization of cementoblast progenitor cells with Bmi-1 and TERT. J Bone Miner Res. 2005;20:50–7.
Komaki M, Iwasaki K, Arzate H, Narayanan AS, Izumi Y, Morita I. Cementum protein 1 (CEMP1) induces a cementoblastic phenotype and reduces osteoblastic differentiation in periodontal ligament cells. J Cell Physiol. 2012;227:649–57.
Hirata A, Sugahara T, Nakamura H. Localization of runx2, osterix, and osteopontin in tooth root formation in rat molars. J Histochem Cytochem. 2009;57:397–403.
Kerr JM, Hiscock DR, Grzesik W, Robey PG, Young MF. The human bone sialoprotein gene contains an NF-E1/YY1 cis-acting sequence with putative regulatory activity. Calcif Tissue Int. 1997;60:276–82.
Chen S, Gluhak-Heinrich J, Martinez M, Li T, Wu Y, Chuang HH, Chen L, Dong J, Gay I, MacDougall M. Bone morphogenetic protein 2 mediates dentin sialophosphoprotein expression and odontoblast differentiation via NF-Y signaling. J Biol Chem. 2008;283:19359–70.
Acknowledgments
We thank Dr. Takeki Tsutsui for suggestions and criticisms throughout this work. This work was supported in part by a Grant-in-Aid for Young Scientists (No. 25862060 to D.T.) from the Japan Society for the Promotion of Science (JSPS) and a Research Grant (2013-2014 to K.K.) from the Nippon Dental University.
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical standards
This study was approved by the Committee of Ethics, the Nippon Dental University School of Life Dentistry at Tokyo (Japan).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Torii, D., Tsutsui, T.W., Watanabe, N. et al. Bone morphogenetic protein 7 induces cementogenic differentiation of human periodontal ligament-derived mesenchymal stem cells. Odontology 104, 1–9 (2016). https://doi.org/10.1007/s10266-014-0182-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10266-014-0182-1