
Abstract
Sagittaria is a genus of ca. 40 species in the aquatic plant family Alismataceae with a nearly global distribution, and a center of diversity in the New World. Two thirds of the known species are native to the Americas, while only a few species are distributed in Africa, Asia and Europe. A previous biogeographic analysis of the genus suggested an African origin for the genus with subsequent dispersal to North America and then to East Asia. Here we expanded the taxon sampling with a focus on the New World taxa and applied species delimitation and biogeographic analyses to revise the knowledge of the phylogeny and evolution of the genus. We obtained largely similar topologies from the chloroplast DNA and nuclear DNA (ITS) data sets. The 74 accessions sampled for our analyses were delimited into 29 species and several cryptic taxa were revealed in widely distributed species. Biogeographic analysis supported basal diversification in South America and subsequent colonization to North America and Asia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aquatic plants are found in aquatic environments throughout the world and tend to show relatively low species diversity and widespread distributions (de Candolle 1855; Sculthorpe 1967). Phenotypic plasticity is often remarkable in aquatic plants and in many cases leads to difficulties in taxonomic and systematic studies (e.g., Kaplan 2002). Recent molecular phylogenetic studies have revealed cases of widespread aquatic plants in which the species diversity had been under-estimated. For example, Ito et al. (2010, 2013, 2015) collected a cosmopolitan species, Ruppia maritima L. (Ruppiaceae), across the world and discerned a species complex of seven taxonomic entities as well as found a cryptic species that was described as a new species from South Africa. Ito et al. (2019) and Li et al. (2020) collected and analyzed multiple samples of Ottelia alismoides (L.) Pers. across Asia and O. ulvifolia (Planch.) Walp. in Africa (Hydrocharitaceae), respectively, and revealed cryptic diversity within these widespread species.
Sagittaria L. is a genus of an aquatic family Alismataceae that contains ca. 40 species in the world. Some species show great phenotypic plasticity depending on the aquatic environment (Haynes and Hellquist 2000). Some Sagittaria species are also widely distributed, such as S. graminea Michx., S. lancifolia L., S. latifolia Willd., and S. montevidensis Cham. and Schltdl in the Americas (Crow and Hellquist 2000; Haynes and Hellquist 2000; Haynes and Holm-Nielsen 1994, 1995; Holm-Nielsen and Haynes 1986; Proctor 2005), S. guayanensis Kunth in the pantropics (Haynes and Hellquist 2000; Wang et al. 2010), and S. natans Pall. in Eurasia (Dandy 1980; Tanaka 2015; Wang et al. 2010). It is possible that these wide ranging Sagittaria species may actually represent species complexes of previously unnoticed diversity.
Biogeography of widely distributed aquatic plants has interested botanists for decades (e.g., Les et al. 2003). A previous study on the phylogeny and biogeography of Alismataceae included 11 Sagittaria accessions representing ten species from Asia and North America and inferred North America and Africa as the ancestral area for the genus (Chen et al. 2012). However, the inference seems preliminary because only a few American species of Sagittaria were sampled, despite the Americas being the center of diversity for the genus with ca. 30 species (Haynes and Hellquist 2000; Holm-Nielsen and Haynes 1986; Keener 2005). In order to address the historical biogeography more accurately, a follow-up analysis based on a taxonomically more comprehensively sampled molecular phylogenetic tree is needed.
In the present study we aim to revisit the phylogeny of Sagittaria by expanding the previously limited taxonomic sampling with a better representation of the species from the Americas. In particular, we collected multiple samples from the widely distributed species mentioned above and applied a species delimitation analysis (STACEY: species tree estimation using DNA sequences from multiple loci; Jones 2017) to test the presence of cryptic species. We also use the new densely sampled molecular phylogeny to revise the biogeographic history of the genus.
Materials and methods
Taxon sampling
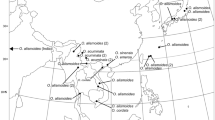
Samples of Sagittaria were collected in the field or obtained from herbarium specimens (Fig. 1; Table 1). The sampled specimens were identified based on the taxonomic treatments of Bogin (1955), Crow and Hellquist (2000), Haynes and Hellquist (2000), and Keener (2005) for the North American species; Holm-Nielsen and Haynes (1986), Haynes and Holm-Nielsen (1994, 1995), and Lehtonen (2018) for the South American species; Wang et al. (2010) and Tanaka (2015) for the Asian species; and Dandy (1980) for European species. Infraspecific taxa in S. graminea and S. montevidensis recognized by Haynes and Hellquist (2000) were treated as species following Keener (2005), namely S. chapmanii (J.G. Sm.) C. Mohr and S. calycina Engelm. Identifications to infraspecific taxa in S. guayanensis followed Haynes and Hellquist (2000) and Wang et al. (2010). A single specimen from Ohio, USA, was tentatively identified as S. cf. australis (J.G. Sm.) Small (OS432686) based on the available digital image of the specimen. Our taxon sampling covered 28 recognized species and two subspecies (Fig. 1; Table 1). Limnophyton Miq. and Caldesia Parl., representatives of two closely related genera of Sagittaria (Chen et al. 2012), were used as outgroup taxa.

Map of sampling localities of Sagittaria species included in this study. Shaded areas correspond to the seven biogeographic areas delimited for the biogeographic analysis. Samples collected in this study are shown with closed dots and those from Chen et al. (2012) with open dots. The number of specimens per species per area is shown in parentheses. Only the native accessions are shown here; the samples of garden origin (one each for S. graminea, S. montevidensis, S. platyphylla, S. potamogetifolia, and S. trifolia) are not shown. The area codes are used for biogeographic analysis (Fig. 4)
DNA extraction, amplification, and sequencing
Total genomic DNA was extracted from silica gel-dried leaf tissue using the CTAB method as described in Ito et al. (2010). Three regions of ptDNA (matK, psbA, and rbcL) and nrITS were PCR amplified with the following primers: matK-390F (Cuénoud et al. 2002) and matK-1520R (Whitten et al. 2000) for matK; Psn3F and Psb3R (Chen et al. 2012) for psbA; rbcL-F1F (Wolf et al. 1994) and rbcL-1379R (Little and Barrington 2003) for rbcL; and ITS-4 and ITS-5 for nrITS (Baldwin 1992). PCR amplification was conducted using TaKaRa Ex Taq polymerase (TaKaRa Bio, Shiga, Japan). PCR cycling conditions were: initial denaturation at 94 °C for 60 s; followed by 30 cycles of 94 °C for 45 s, 52 °C for 30 s, 72 °C for 60 s, with a final extension of 72 °C for 5 min. PCR products were cleaned using ExoSAP-IT purification (GE Healthcare, Piscataway, New Jersey), and then amplified using Big Dye Terminator ver. 3.1 (Applied Biosystems, Foster City, California) using the same primers as those used for the PCR amplifications. DNA sequencing was performed with a 3130xl Genetic Analyzer (Applied Biosystems). Automatic base-calling was verified by eye in Genetyx-Win ver. 3 (Software Development Co., Tokyo, Japan). All sequences generated in the present study have been submitted to the DNA Data Bank of Japan (DDBJ), which is linked to GenBank. The accession numbers and voucher specimen information are presented in Table 1.
Molecular phylogenetic analysis
Sequences were aligned using Mafft ver. 7.058 (Katoh and Standley 2013) and then inspected manually. Indels were not coded because length variations were either ambiguous (nrITS) or observed only in S. graminea GH215 (a 9 bp-long insertion in matK). Accessions for ten Sagittaria spp. generated by Chen et al. (2012) were downloaded and included in our data sets except for S. isoetiformis, for which the ITS sequence (JF780976) apparently represents a contaminated sample or submission error, being identical to a sequence of S. latifolia (JF780975). Phylogenetic analyses were independently performed for ptDNA (matK, psbA, and rbcL) and nrITS data sets to identify possible incongruence between the different genomic regions. All 74 ingroup and the two outgroup accessions were included in the ptDNA data set, whereas the Caldesia outgroup sequence was excluded from the fast-evolving nrITS data set to allow more accurate alignment.
Phylogenies were reconstructed using maximum parsimony (MP), maximum likelihood (ML), and Bayesian inference (BI; Yang and Rannala 1997). In the MP analysis, conducted in PAUP* ver. 4.0b10 (Swofford 2002), a heuristic search was performed with 100 random addition replicates and tree-bisection-reconnection (TBR) branch swapping, with the MulTrees option in effect. The MaxTrees option was set at 100,000. Bootstrap analyses (Felsenstein 1985) were performed using 1,000 replicates with TBR branch swapping and simple addition sequences. The MaxTrees option was set at 1,000 to avoid entrapment in local optima. For the ML analysis, we used the RAxML BlackBox online server (Kozlov et al. 2019), which supports GTR-based models of nucleotide substitution (Stamatakis 2006). The maximum likelihood search option was used to find the best-scoring tree after bootstrapping. The gamma model of rate heterogeneity was applied as default. Statistical support for branches was calculated by rapid bootstrap analysis of 100 replicates (Stamatakis et al. 2008).
BI analyses were conducted with MrBayes ver. 3.2.2 (Ronquist and Huelsenbeck 2003; Ronquist et al. 2012) run on the CIPRES portal (Miller et al. 2010) after the best models had been determined in MrModeltest ver. 3.7 (Nylander 2002); these models were GTR + I + G and HKY + G for ptDNA and nrITS data sets, respectively. Analyses were run for 3,000,000 generations for ptDNA and nrITS data sets, respectively, sampling every 1,000 generations and discarding the first 25% as burn-in. The convergence and effective sampling sizes (ESS > 200 of all the parameters) were checked in Tracer ver. 1.6 (Rambaut et al. 2014). All trees were visualized using FigTree ver. 1.3.1 (Rambaut 2009). Nodes were recognized as strongly (≥ 90% MP bootstrap support (BS), ≥ 90% ML BS and ≥ 0.95 posterior probability (PP)), moderately (≥ 70% MP BS, ≥ 70% ML BS and ≥ 0.9 PP), or weakly (< 70% MP BS, < 70% ML BS and < 0.9 PP) supported. The data matrices and the MP, RAxML and BI trees were deposited in Treebase (TB2:S26217).
Molecular species delimitation
A Bayesian coalescent method of species delimitation was performed using STACEY (Jones 2017), which is an extension of DISSECT (Jones et al. 2015). Species tree estimation was performed in STACEY and species delimitation was conducted in SpeciesDelimitationAnalyser (Jones et al. 2015; Jones 2017). STACEY was implemented in BEAST ver. 2.4.4 (Bouckaert et al. 2014; Drummond and Rambaut 2007; Drummond et al. 2006). We ran STACEY using a multilocus data set (ptDNA and nrITS) with all ingroup species; outgroup species were excluded to avoid rate differences and hidden substitutions between ingroup and outgroup species (Bengt Oxelman, personal communication, November 22, 2016). We performed two independent runs of MCMC chains with ten million generations, sampling every 1,000 generations. Convergence of the stationary distribution was checked by visual inspection of plotted posterior estimates using Tracer ver. 1.6 (Rambaut et al. 2014). After discarding the first 1,000 trees as burn-in, the samples were summarized in the maximum clade credibility tree using TreeAnnotator ver. 1.6.1 (Drummond and Rambaut 2007) with a posterior probability limit of 0.5 and summarizing of mean node heights. The results were visualized using FigTree ver. 1.3.1 (Rambaut 2009).
Species tree and biogeographic analyses
Biogeographical reconstructions were performed using the species tree generated by BEAST. STACEY was run using a multilocus dataset (ptDNA and ITS) utilizing all 74 ingroup terminals and the outgroups assigned to 31 operational taxonomic units (OTUs), 29 of which were retrieved as species or minimal clusters in the STACEY analysis. The furthest outgroup was trimmed using RASP v.4.2 (Yu et al. 2020). Reconstruction of historical biogeography was performed using BioGeoBEARS (Matzke 2013) implemented in RASP v.4.2 (Yu et al. 2020). Model fit was assessed using the Akaike information criterion and likelihood-ratio tests. “Max areas” was set to the default (two). The following six biogeographic areas were defined: (A) Europe; (B) Africa; (C) temperate Asia; (D) tropical Asia; (E) North America; (F) South America (Fig. 1). Central America is included in North America because no species endemic to Central America are known and some North American species extend to Mexico and the West Indies, i.e., S. demersa J.G. Sm., S. graminea, and S. longiloba Engelm. ex J.G. Sm. (Haynes and Hellquist 2000). Sagittaria longiloba, previously known from North and Central America (Nicaragua) (Haynes and Hellquist 2000), was coded to be distributed in North and South America because one of our samples (SL466) was collected from Venezuela (Fig. 1, Table 1).
Results
Molecular phylogeny
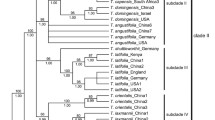
The ptDNA data set comprising three genes included 3,107 aligned characters, of which 201 were parsimony-informative. Analysis of this data set overflowed the imposed limit of 100 000 MP trees (tree length = 511 steps; consistency index = 0.77; retention index = 0.90). The strict consensus of these trees, the RAxML tree, and the MrBayes 50% majority rule consensus tree showed no strongly supported incongruences; thus, only the Bayesian tree is presented here (Fig. 2a). The nrITS data set included 751 aligned characters, of which 259 were parsimony-informative. Analysis of this data set overflowed the imposed limit of 100,000 MP trees (tree length = 668 steps; consistency index = 0.71; retention index = 0.91). The strict-consensus of these trees, the ML tree and the Bayesian 50% majority rule consensus tree showed no incongruent phylogenetic relationships; thus, only the MrBayes tree is presented here (Fig. 2b).

MrBayes trees of Sagittaria based on a ptDNA and b nrITS datasets. Samples collected in this study are associated with the specified vouchers. Branch lengths are proportional to the number of substitutions per site as measured by the scale bar. Values above the branches represent the maximum parsimony and maximum likelihood bootstrap support values (MP BS/ML BS), and Bayesian posterior probabilities (PP). BS < 70% and PP < 0.9 are indicated by hyphens while those of ≥ 90% and ≥ 0.95 are shown as asterisks. Well-supported clades are highlighted by gray rectangles with numbers
The monophyly of the genus Sagittaria was highly supported (98% MP BS, 93% ML BS, 1.0 PP in Fig. 2a). While Sagittaria guayanensis (clade 1) was weakly resolved as sister to the rest of the genus in ptDNA with S. planitiana G. Agostini (clade 2) resolved as the sister to the rest of the species, the two species were clustered in nrITS (84% MP BS, 100% ML BS, 1.0 PP) and resolved together as sister to all other Sagittaria. Clade 5 plus S. montevidensis SL180 and S. sprucei Micheli were resolved as sister to clades 6–22 in both ptDNA and nrITS (98% MP BS, 100%ML BS; 1.0 PP in Fig. 2a; 99% MP BS, 100% ML BS; 1.0 PP in Fig. 2b). Sagittaria longiloba was weakly clustered with clades 6–14 in ptDNA (0.97 PP in Fig. 2a) but resolved as sister to clades 16–22 in nrITS (87% MP BS, 91% ML BS, 1.0PP in Fig. 2b). While clades 6–9 were weakly supported as monophyletic in ptDNA (56% MP BS, 70% ML BS, 1.0 PP in Fig. 2a), clades 7–9 were resolved with clades 10–14 in nrITS (100% MP BS, 96% ML BS, 1.0PP in Fig. 2b). Clades 16–18 formed a monophyletic group in both analyses (100% MP BS, 100% ML BS, 1.0 PP in both Fig. 2a, b) and were resolved as sister to clades 19–22 (76% MP BS, 81% ML BS, 1.0 PP in Fig. 3a; 100% MP BS, 97% ML BS, 1.0 PP). Sagittaria sagittifolia L. was weakly clustered with clade 19 in ptDNA (< 50% MP BS, < 70%ML BS; 1.0 PP in Fig. 2a) but was clustered with clades 21–22 in nrITS (75% MP BS, 92% ML BS, 0.98 PP in Fig. 2b).

Maximum sum of clade credibility SMC-tree based on multilocus (ptDNA and nrITS) data set of Sagittaria from BEAST 2 analysis and similarity matrix from STACEY analysis. Posterior probabilities from BEAST 2 are given for major clades on the branches. The squares in the matrix represent posterior probabilities (white = 0, black = 1) for pairs of individuals belonged to the same cluster. The horizontal lines represent species boundaries suggested by STACEY analysis. Taxa that receive taxonomic changes are shown with parentheses (see “Discussion” for more detail)
Species delimitation using STACEY
SpeciesDelimitationAnalyser generated 1,527 patterns of clusters from the MCMC runs. Classifications with 28 or fewer Sagittaria minimal clusters (species) received higher posterior probabilities (Table S1). Under these classifications, the species delimitation among S. latifolia and S. australis as well as the status of the two subspecies of S. guayanensis remained unclear. The similarity scores among these clusters seem to be low enough to support distinct species, i.e., the similarity score of S. australis vs. S. latifolia SL411 was 0.26–0.27; S. latifolia SL411 vs. S. latifolia excluding SL411 was 0.24–0.42; S. australis vs. S. latifolia excluding SL411 was 0.43–0.60; S. guayanensis subsp. guayanensis vs. S. guayanensis subsp. lappula was 0.31 (Table S2). Therefore, for further analyses we recognized the two subspecies of S. guayanensis, S. australis, S. latifolia and S. latifolia SL411 as species. Hence, a total of 29 species with the posterior probability of 0.10 PP was accepted (Fig. 3).
Biogeography
The BioGeoBEARS tool in RASP assigned the DEC + j scenario as the best model (AICc_wt = 0.52), followed by DIVALIKE + J (AICc_wt = 0.35) and BAYAREALIKE + J (AICc_wt = 0.13). Under the DEC + j scenario, the ancestral area of the root of Sagittaria (node I) was resolved as South America (0.65 PP) (node I in Figs. 4, S1). The ancestral area of node II was South America (0.71 PP) (Figs. 4, S1). The ancestral area of node III was North America (0.55 PP) (Figs. 4, S1). The ancestral area for the clade containing Asian species and S. latifolia (node IV) was North America (0.59 PP). The ancestral area for Asian species (node V) was tropical Asia (0.54 PP), or temperate Asia (0.34 PP). The ancestral area for S. calycina, S. intermedia, and S. montevidensis (node VI) was South America (0.80 PP). The ancestral area for the two subspecies of S. guayanensis (node VII) was North and South America (0.61 PP).

Ancestral range reconstruction on the phylogenetic tree of Sagittaria with the best fitting DEC + j model in BioGeoBEARS. The branch width and color indicate posterior priorities (thick black lines with higher probabilities). The areas inferred with the highest posterior probabilities are shown; those which have similar posterior probabilities for different areas inferred are shown below. The dotted arrows indicate colonization events out of South America. The closed squares on the right indicate distribution areas for each species. Area codes are as follows: a Europe, b Africa, c Temperate Asia, d Tropical Asia, e North America, f South America. The roman numerals at nodes are mentioned in the text. The outgroup was trimmed
Discussion
Phylogeny of Sagittaria
Molecular phylogenetics of Sagittaria have been previously studied by Keener (2005), who sampled the 5S-NTS marker for 73 accessions representing 38 taxa, and Chen et al. (2012) in their phylogenetic study of Alismataceae by using nrITS and ptDNA (matK, psbA, and rbcL) with a set of 10 Sagittaria species. Here, we gathered 74 Sagittaria samples, including eight samples from Keener (2005), three from Lehtonen and Myllys (2008), and nine from Chen et al. (2012), and conducted the most comprehensive molecular phylogenetic analysis of the genus so far. The results showed no significant topological incongruence between the previous results, i.e., (i) S. guayanensis and S. montevidensis were among the early diverged species (clades 1–5 in Fig. 2; Chen et al. 2012; Keener 2005); (ii) a clade containing S. potamogetifolia Merr. and S. pygmaea Miq. was recovered (clades 16–18 in Fig. 2; Chen et al. 2012; Keener 2005); (iii) a clade containing S. latifolia, S. natans, and S. trifolia was recovered (clades 19–22 in Fig. 2; Chen et al. 2012; Keener 2005, Fig. 1a, b except S. isoetiformis for which the ITS sequence apparently represents an error, being identical to a sequence of S. latifolia).
Cryptic diversity in Sagittaria
Sagittaria montevidensis is currently considered as a widely distributed species ranging from North America to Mexico and South America (Haynes and Hellquist 2000). Keener (2005) divided this species of four subspecies into two species, each having two subspecies, i.e., S. calycina in North and Central America, and S. montevidensis in South America. Sagittaria intermedia from Central America is also clustered with these two species in our molecular phylogeny. Given the morphological similarity, S. sprucei from South America was suggested to be related to these species (Keener 2005) and our phylogeny confirmed this.
The South American samples identified as S. montevidensis were split so that the Bolivian sample (SL180) was resolved highly distinct from the others (YI2030 from Argentina, SL258 from Paraguay, and two garden-origin). Two varieties of S. montevidensis are generally recognized: var. chilensis from temperate South America and var. montevidensis from tropical South America (Bogin 1955; Keener 2005). They can be distinguished by the absence or smaller size of the basal lobes, and by the carpellate pedicels being inflated at the base of the flowers in var. chilensis, whereas in var. montevidensis the basal lobes are longer than or equal to the remainder of the blade, and pedicels of carpellate flowers are merely thickened at the base of the flowers. The sample from Bolivia had large lobes and merely thickened pedicels and therefore cannot represent var. chilensis. In addition, it lacked the purple spots at the base of the petals, a character typical for S. montevidensis, but instead had yellow spots. However, the current sampling and limited herbarium material available to us does not allow well justified taxonomic conclusions at the moment, but further taxonomic studies in the South American S. montevidensis are clearly needed to clarify the species delimitation within this apparent species complex.
Sagittaria latifolia is currently treated as containing two varieties (var. latifolia and var. pubescens) distinguished by the presence or absence of pubescence over the plant (Bogin 1955). While Ecuadorian specimens were identified as S. latifolia var. latifolia (Holm-Nielsen and Haynes 1986), recent floras of North America, i.e., Crow and Hellquist (2000), Haynes and Hellquist (2000), and Keener (2005), tend not to recognize the varieties, adding that “many (specimens) from the southeastern United States are pubescent” (Haynes and Hellquist 2000). We found no significant genetic differences among the North and South American samples, whereas the species delimitation analysis distinguished—although weakly—the Mexican sample (SL411) from the other North and South American samples (Fig. 3). Sculthorpe (1967) pointed out the presence of sporadic populations of S. latifolia in Central America which are discontinuous from the main distribution area in North America, indicating geographic isolation in Mexican populations including our sample. The broad and somewhat discontinuous distribution of the S. latifolia complex is a subject for future taxonomic studies.
Evidence for colonization out of South America
Chen et al. (2012) resolved the paleotropical clade composed of Astonia S.W.L. Jacobs, Limnophyton, and Wiesneria Micheli as the sister to Sagittaria and inferred Africa and North America as ancestral areas for Sagittaria. This contrasts with our result of a South American origin for the genus. Unlike Chen et al. (2012), we included only one outgroup, Limnophyton, in our biogeographic analysis and the results may have been different if a broader taxonomic sampling had been used. However, the South American Sagittaria are much better sampled in our study compared to Chen et al. (2012), and their phylogenetic position clearly indicates that the deepest split in the genus occurred in South America. It therefore seems reasonable to assume that the diversification of modern Sagittaria began in South America, even if the ancestor had reached South America by dispersal from Africa.
Our analysis revealed multiple events of colonization out of South America, leading to (i) the first invasion to North America (node III in Fig. 4); (ii) the second invasion to North America at the most recent common ancestor for S. calycina and its allies (although the clade of S. calycina and S. intermedia is less-supported); and (iii) a range expansion of the most recent common ancestor for S. guayanensis and its allies to North America and a subsequent intercontinental dispersal of S. (guayanensis subsp.) lappula to paleotropics (Fig. 4). Subsequent speciation occurred in North America, where at least ten species evolved (Fig. 4). Furthermore, we infer the range expansion from North America to East Asia at node V and another dispersal event from North America to Europe with a subsequent range expansion to Asia, resulting in S. sagittifolia, S. natans and S. trifolia (Fig. 4).
Some Sagittaria species are widely distributed in the Americas, such as S. lancifolia, S. latifolia, and S. longiloba (Haynes and Hellquist 2000; Holm-Nielsen and Haynes 1986). Our biogeographic analysis showed North America for the most recent common ancestors for these species and their sister species (Fig. 4), suggesting that, at species level, range expansions from North America back to South America occurred at least three times.
Implication for taxonomy and evolution of Sagittaria species
Sagittaria guayanensis is widespread in most tropical countries of the world and is divided into two subspecies: S. guayanensis subsp. guayanensis in Neotropics (Western hemisphere) and S. guayanensis subsp. lappula in Paleotropics (Eastern hemisphere) (Haynes and Hellquist 2000; Holm-Nielsen and Haynes 1986). The monophyly of S. guayanensis was confirmed by Keener (2005) as well as our ptDNA and nrITS phylogenies (Fig. 2). However, our species delimitation analysis suggested that the subspecies could be recognized as two different species (Fig. 3). We therefore recommend restoring species status of S. guayanensis subsp. lappula as S. lappula D. Don.
Sagittaria filiformis, S. kurziana, and S. subulata are morphologically highly similar, narrowly distributed endemics to southeastern North America (Haynes and Hellquist 2000). Keener (2005) as well as we found no, or very limited genetic variation among these taxa (Fig. 2). We therefore propose treating these taxa as varieties of S. subulata: Sagittaria subulata var. gracillima (S. Watson) J.G. Sm., Sagittaria subulata var. kurziana (Glück) Bogin, and Sagittaria subulata var. subulata, respectively.
Conclusions
We provided new phylogenetic insights into the diversity and biogeography of Sagittaria, a nearly cosmopolitan genus with greatest diversity in the Americas. Our molecular phylogenetic analyses based on ptDNA and nrITS data sets using an expanded taxon sampling revealed cryptic diversity in widely distributed S. latifolia and S. montevidensis. A previously inferred African–North American origin of the genus was revised to an ancestral distribution in South America, with deepest diversifications occurring there subsequently followed by speciation in North America. Three colonization events occurred out of South America, towards North America and Asia. At species level, S. lancifolia, S. latifolia, and S. longiloba represent range expansions back to South America. Eurasia has been occupied twice, possibly by means of relatively short-distance circumpolar dispersal from North America. The biogeographic insights revealed in this study support the importance of inter-continental dispersal in widely distributed aquatic plants. Our study demonstrates the importance of broad sampling of widespread species for addressing cryptic diversity in aquatic plants.
References
Baldwin BG (1992) Phylogenetic utility of the internal transcribed spacers of nuclear ribosomal DNA in plants: an example from the Compositae. Mol Phylogenet Evol 1:3–16
Bogin C (1955) Revision of the genus Sagittaria (Alismataceae). Mem N Y Bot Gard 9:179–223
Bouckaert RR, Heled J, Kühnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537
Chen LY, Chen JM, Gituru RW, Temam TD, Wang QF (2012) Generic phylogeny and historical biogeography of Alismataceae, inferred from multiple DNA sequences. Mol Phylogenet Evol 63:407–416
Crow GE, Hellquist CB (2000) Aquatic and wetland plants of northeastern North America. Two Monocotyledons: Angiosperms. University of Wisconsin Press, Madison
Cuenoud P, Savolainen V, Chatrou LW, Powell M, Grayer RJ, Chase MW (2002) Molecular phylogenetics of Caryophyllales based on nuclear 18S rDNA and plastid rbcL, atpB, and matK DNA sequences. Am J Bot 89:132–144
Dandy JE, Sagittaria L (1980) Flora Europaea. In: Tutin TG, Heywood VH, Burges NA, Moore DM, Valentine DH, Walters SM, Webb DA, Chater AO, Richardson IBK (eds) Alismataceae to Orchidaceae, vol 5. Cambridge University Press, Cambridge, pp 1–2
de Candolle A (1855) Gèographie botanique raisonnèe; ou, exposition des faits principaux et des lois concernant la distribution gèographique des plantes de l’ èpoque actuelle, vol 2. Victor Masson, Paris, p 1365
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214–222
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Haynes RR, Hellquist CB (2000) Alismataceae. In: Flora of North America Editorial Committee (Eds.). Flora of North America North of Mexico. vol 22. Oxford University Press, Oxford, pp 7−25
Haynes RR, Holm-Nielsen LB (1994) The Alismataceae. Flora neotropica. Monogr 64:1–112
Haynes RR, Holm-Nielsen LB (1995) Alismataceae. In: Berry PE, Holst BK, Yatskievych K (eds) Flora of venezuelan guayana, vol 2. Missouri Botanical Garden Press, St. Louis, pp 377–383
Holm-Nielsen LB, Haynes RR (1986) 191. Alismataceae. In: Harling G, Andersson L (eds) Flora of ecuador, vol 26. Publishing House of the Swedish Research Councils, Stockholm, pp 1–24
Ito Y, Ohi-Toma T, Murata J, Tanaka N (2010) Hybridization and polyploidy of an aquatic plant, Ruppia (Ruppiaceae), inferred from plastid and nuclear DNA phylogenies. Am J Bot 97:1156–1167
Ito Y, Ohi-Toma T, Murata J, Tanaka N (2013) Comprehensive phylogenetic analyses of the Ruppia maritima complex focusing on taxa from the Mediterranean. J Plant Res 126:753–762
Ito Y, Tanaka N, Ohi-Toma T, Murata J, Muasya AM (2015) Phylogeny of Ruppia (Ruppiaceae) revisited: molecular and morphological evidence for a new species from Western Cape, South Africa. Syst Bot 40:942–949
Ito Y, Tanaka N, Barfod AS, Bogner J, Li J, Yano O, Gale SW (2019) Molecular phylogenetic species delimitation in the aquatic genus Ottelia (Hydrocharitaceae) reveals cryptic diversity within a widespread species. J Plant Res 132:335–344
Jones GL (2017) Algorithmic improvements to species delimitation and phylogeny estimation under the multispecies coalescent. J Math Biol 74:447–467
Jones GL, Aydin Z, Oxelman B (2015) DISSECT: an assignment-free Bayesian discovery method for species delimitation under the multispecies coalescent. Bioinformatics 31:991–998
Kaplan Z (2002) Phenotypic plasticity in Potamogeton (Potamogetonaceae). Folia Geobot 37:141–170
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Keener BR (2005) Molecular systematics and revision of the aquatic monocot genus Sagittaria (Alismataceae). Dissertation, University of Alabama
Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A (2019) RAxML-NG: a fast, scalable, and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics, btz305
Lehtonen S (2018) Alismataceae. In: Ramella L (ed) Flora del paraguay, vol 49. Conservatoire et Jardin botaniques de la Ville de Genève, Geneve, pp 1–42
Lehtonen S, Myllys L (2008) Cladistic analysis of Echinodorus (Alismataceae): simultaneous analysis of molecular and morphological data. Cladistics 24:218–239
Les DH, Crawford DJ, Kimball RT, Moody ML, Landolt E (2003) Biogeography of discontinuously distributed hydrophytes: a molecular appraisal of intercontinental disjunctions. Intl J Plant Sci 164:917–932
Li Z, Ngarega BK, Lehtonen S, Gichira AW, Karichu MJ, Wang QF, Chen JM (2020) Cryptic diversity within the African aquatic plant Ottelia ulvifolia (Hydrocharitaceae) revealed by population genetic and phylogenetic analyses. J Plant Res 133:373–381
Little DP, Barrington DS (2003) Major evolutionary events in the origin and diversification of the fern genus Polystichum (Dryopteridaceae). Am J Bot 90:508–514
Matzke NJ (2013) Probabilistic historical biogeography: new models for founder-event speciation, imperfect detection, and fossils allow improved accuracy and model-testing. Front Biogeogr 5:242–248
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the gateway computing environments workshop (GCE), 14 Nov 2010, New Orleans, pp 1–8. http://www.phylo.org/sub_sections/portal/cite.php
Nylander JAA (2002) MrModeltest. Ver. 1.0. Program distributed by the author. Department of Systematic Zoology, Uppsala University, Uppsala. Retrieved from https://www.ebc.uu.se/systzoo/staf/nylander.html
Proctor GR (2005) Family 4. Alismataceae water-plantain family. In: Acevedo-Rodriguez P, Strong MT (eds) Monocotyledons and gymnosperms of Puerto Rico and the Virgin Islands. Contributions from the United States national Herbarium, vol 52. Department of Botany, National Museum of Natural History, Washington, pp 58–63
Rambaut A (2009) FigTree ver. 1.3.1: Tree Figure Drawing Tool. Retrieved from https://tree.bio.ed.ac.uk/software/fgtree/. Accessed 4 Jan 2011
Rambaut A, Suchard MA, Xie W, Drummond AJ (2014) Tracer. ver. 1.6. Retrieved from https://beast.bio.ed.ac.uk/Tracer. Accessed 8 Sept 2018
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efcient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Sculthorpe CD (1967) The biology of aquatic vascular plants. Edward Arnold, London
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihoodbased phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57:758–771
Swoford DL (2002) PAUP: Phylogenetic analysis using parsimony (and other methods). ver. 40b10. Sinauer Associates, Sunderland
Tanaka N (2015) Alismataceae. In: Ohashi H, Kadota Y, Murata J, Yonekura K, Kihara H (eds) Wild flowers of Japan, vol 1. Heibonsha, Tokyo, pp 115–117 ((in Japanese))
Wang QF, Guo YH, Haynes RR, Hellquist CB (2010) Alismataceae. In: Wu C-Y, Raven PH, Hong D-Y (eds) Flora of China, vol 23. Science Press. Beijing & Missouri Botanical Garden Press, St. Louis, pp 84–89
Whitten WM, Williams NH, Chase MW (2000) Subtribal and generic relationships of Maxillarieae (Orchidaceae) with emphasis on Stanhopeinae: combined molecular evidence. Am J Bot 87:1842–1856
Wolf PG, Soltis PS, Soltis DE (1994) Phylogenetic relationships of Dennstaedtioid ferns: evidence from rbcL sequences. Mol Phylogenet Evol 3:383–392
Yang Z, Rannala B (1997) Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo method. Mol Biol Evol 14:717–724
Yu Y, Blair C, He XJ (2020) RASP 4: ancestral state reconstruction tool for multiple genes and characters. Mol Biol Evol 37:604–606
Acknowledgements
The authors thank Drs. P. G. Aceñolaza (Buenos Aires), M. Sasagawa (Yamagata), and O. Yano (Okayama) for assistance in the field; D. Boufford and E. Wood (GH) and J. V. Freudenstein (OS) for hospitality during the first author’s recent visits; and C. Ishii (Tsukuba) for help with DNA sequencing. This research was partly supported by the international cooperative project “Biological inventory with special attention to Myanmar: Investigations of the origin of southern elements of Japanese flora and fauna” as the integrated research initiated by the National Museum of Nature and Science, Japan based on MoU between Forest Department, Ministry of Natural Resources and Environmental Conservation, Myanmar to NT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ito, Y., Tanaka, N., Keener, B.R. et al. Phylogeny and biogeography of Sagittaria (Alismataceae) revisited: evidence for cryptic diversity and colonization out of South America. J Plant Res 133, 827–839 (2020). https://doi.org/10.1007/s10265-020-01229-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10265-020-01229-5