
Abstract
To investigate the phylogeography of the Japanese pale chub Opsariichthys platypus, we examined the genetic differentiation, phylogenetic relationships, and historical demography using mitochondrial cytochrome b gene sequences of 788 O. platypus from 124 localities throughout the Japanese Archipelago. Molecular phylogenetic analyses revealed that Japanese O. platypus is divided into three major clades (western Japan: WJ; eastern Japan: EJ; and Kyushu: KY), and that among these clades, KY is remarkably differentiated from the others. The distribution of the EJ and KY clades clearly reflects their respective geographical ranges: the EJ and KY clades are restricted to the eastern region of the Ibuki–Suzuka Mountains in central Honshu and northern Kyushu, respectively. In contrast to the EJ and KY clades, the WJ clade is widely distributed throughout the Japanese Archipelago, including areas where O. platypus is not naturally distributed (e.g., northern part of Honshu, southern Kyushu, and Tokunoshima). In addition, nearly all the WJ haplotypes in the non-indigenous regions were the same as or similar to the haplotypes in Lake Biwa, and the WJ haplotypes in the distribution ranges of the EJ and KY clades were also same as or similar to those of Lake Biwa, indicating that the distribution of WJ clade had been the result of inadvertent releases in conjunction with releases of Plecoglossus altivelis from Lake Biwa. The estimated divergence time indicated that each clade was formed during or before the Pleistocene, and mismatch distribution test suggested the occurrence of the population expansion in three clades and the time since expansion was 120,000–226,000 years. This study demonstrates that O. platypus exhibits clear genetic differentiation among regional populations, and that range expansion following divergence caused by uplifting of mountains is important for distribution and genetic structuring of O. platypus. In addition, artificial introductions of non-indigenous populations have proceeded throughout the Japanese Archipelago, indicating ongoing loss of the genetic features due to introductions or introgression. Further efforts should be made toward a comprehensive understanding of the current state of introductions of non-indigenous O. platypus populations and the potential influence of invasive populations, including loss of genetic diversity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
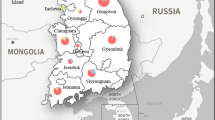
The Japanese Archipelago extends approximately 3000 km from north to south and consists of four major islands and many small islands (Fig. 1). The geography and climate of the archipelago varies widely from subarctic to subtropical (Biodiversity Center of Japan 2010). The many straits and mountain ranges, which began forming and/or uplifting during the Pliocene–early Pleistocene (Yonekura et al. 2001; Machida et al. 2001, 2006), serve as long-term biogeographical barriers to migration and dispersal of animals and plants. These variations in geography and climate, including periodic glacial–interglacial cycles, have played important roles in creating regional faunas and floras and high levels of biodiversity within the archipelago (e.g., Oba 1994; Millien-Parra and Jaeger 1999; Ota 2000). Studies of the geographic patterns of genetic features have also offered insights into the roles of complex geographic history in rich biodiversity and high levels of endemism in the archipelago (e.g., mammals: Ohnishi et al. 2009; Nunome et al. 2010; amphibians: Nishizawa et al. 2011; Honda et al. 2012; Tominaga et al. 2013; arthropods: Koizumi et al. 2012; Schoville et al. 2013; Suzuki et al. 2014).

a Sampling localities of Opsariichthys platypus in Japan. Detailed information on each locality is shown in ESM Table S1. b The assumed natural range of O. platypus before 1912. This map is cited from Mizuguchi (1990)
Freshwater fish faunas are strongly affected by geography, climate, and historical processes. Several Japanese fish species, for example, exhibit high genetic fragmentation or divergence associated with geography, even at small spatial scales (e.g., Takehana et al. 2003; Watanabe and Mori 2008; Tominaga et al. 2009, 2016), reflecting historical geographic events within the Japanese Archipelago. In addition, low altitude areas within the Japanese Archipelago were never covered by ice during glacial periods (Yonekura et al. 2001), and, in consequence, the primary freshwater fish fauna has developed continuously since the archipelago was formed (Watanabe et al. 2006). Therefore, studies focused on the biogeography and/or phylogeography of freshwater fish can provide valuable insights into the historical processes responsible for the high levels of endemism and biodiversity within the archipelago. However, although phylogeographic studies have been conducted on several fish species, for example Lefua echigonia (Saka et al. 2003), Misgurnus anguillicaudatus (Morishima et al. 2008), Cobitis biwae (Kitagawa et al. 2003), Pseudogobio esocinus (Tominaga et al. 2016), Oryzias latipes (Matsuda et al. 1997; Takehana et al. 2003), Lethenteron spp. (Yamazaki et al. 2003), Liobagrus reinii (Nakagawa et al. 2016), Hemigrammocypris rasborella (Watanabe et al. 2014), Cyprinus carpio (Mabuchi et al. 2008), Biwia zezera (Watanabe et al. 2010b), Niwaella delicata (Kitagawa et al. 2001), and Gnathopogon elongatus (Kakioka et al. 2013), the observed phylogeographic patterns of these species were not concordant. For instance, some studies revealed that large genetic divergence could be attributed to the uplifting of the Fossa Magna (e.g., Kitagawa et al. 2003; Saka et al. 2003; Mabuchi et al. 2008; Nakagawa et al. 2016), while others did not show similar divergence pattern (e.g., Matsuda et al. 1997; Takehana et al. 2003; Morishima et al. 2008). In addition, the Ibuki–Suzuka Mountains could be a primary factor for genetic divergence in some species (e.g., Watanabe et al. 2010b, 2014), but not in others (e.g., Kitagawa et al. 2001; Kakioka et al. 2013; Nakagawa et al. 2016). These facts suggest that general pattern of genetic divergence and their relation to geographical patterns have not been well understood. Thus, further study is needed to understand the geographical factors underlying the formation of the Japanese freshwater fish fauna.
The pale chub Opsariichthys platypus (formerly called Zacco platypus in many literatures) is a cyprinid fish distributed in East Asia, including China, Korea, and Japan, and one of the most common freshwater fish in this area. In Japan, O. platypus is found in temperate rivers in the western region of Japan (Kyushu, Shikoku, and western part of Honshu) and is one of the most common species in this region (Hosoya 2013). Such a wide distribution may indicate that their genetic variation reflects historical geographic events throughout the Japanese archipelago, and, thus, a study of its genetic structure provides good opportunities for investigating the processes by which freshwater fish faunas have been established within the Japanese Archipelago. However, genetic divergence and its geographical variation have never been studied in this species, and phylogeographic pattern is not known. In addition, the range of O. platypus has expanded across the whole of Japan, probably due to inadvertent releases in conjunction with releases of Plecoglossus altivelis from Lake Biwa, which have been conducted since 1924, as well as subsequent releases of such introduced O. platypus to other rivers for fisheries improvement and angling (Fig. 1b; Mizuguchi 1990). As a result, the range of O. platypus now includes northern Honshu and Hokkaido (Mizuguchi 1990). Such anthropogenic introductions, as well as a lack of information on phylogenetic relationships, have hampered the efforts to clarify the process by which genetic divergences were geographically constructed.
In this study, we examined the genetic differentiation, phylogenetic relationships, and historical demography of O. platypus using mitochondrial cytochrome b (cyt b) gene sequences to investigate the detailed phylogeography and genetic relationships among O. platypus populations throughout Japan, and to understand their relationship with the geological history of the Japanese Archipelago.
Materials and methods
Sample collection. Using hand nets, casting nets, and fishing cages, we collected 788 specimens of Opsariichthys platypus from 124 localities between 2006 and 2009, covering its whole range in the Japanese Archipelago (Fig. 1). Fin samples or whole body samples were preserved in 99 % ethanol.
DNA extraction, amplification, and alignment. Total genomic DNA was extracted from skeletal muscle samples or fin samples using a DNeasy Blood & Tissue Kit (QIAGEN, Venlo, Netherlands), a High Pure PCR Template Preparation Kit (Roche Diagnostic, Basel, Switzerland), or a ChargeSwitch gDNA Mini Tissue Kit (Life Technologies, Carlsbad, CA, USA), according to the manufacturer’s instructions. Total genomic DNA was used to amplify DNA fragments by polymerase chain reaction (PCR). For PCR amplification of cyt b, the following primers were used: L14690-Cb-AH, 5′-GGT CAT AAT TCT TGC TCG GA-3′, and H15913-Thr-AH, 5′-CCG ATC TTC GGA TTA CAA GAC CG-3′. The PCR was carried out using an automated thermal cycler (2720 Thermal Cycler; Applied Biosystems, Foster City, CA, USA) in 10 µl of the reaction mixture containing 0.625 units of Taq polymerase (TaKaRa Ex Taq; TaKaRa, Otsu, Japan), 0.125 µM of each primer, 0.25 mM dNTP, 2.0 µl of Crimson Taq Reaction Buffer (New England Biolabs, Ipswich, MA, USA), and 1.0 µl of genomic DNA as a template. The thermal conditions were as follows: initial denaturation for 1 min at 95 °C, 30 cycles of incubation for 1 min at 95 °C, 1 min at 55 °C, and 2 min at 72 °C, and hold at 10 °C. The PCR products were purified using an Exo SAP-IT Kit (GE Healthcare, Buckinghamshire, England). The purified DNA products were directly sequenced using the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit ver. 3.1 in an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). The sequence data of 1004 bp of partial cyt-b were aligned with CLUSTAL X ver. 2.0 (Larkin et al. 2007) to identify nucleotide variations and defining haplotypes. We also used sequence data from the DNA databank DDBJ/EMBL/GenBank [accession number: AB198972 in Sasaki et al. 2007; Electronic Supplementary Material (ESM) Table S1].
Phylogenetic analyses and estimating divergence time. Phylogenetic analyses were performed by the neighbor-joining (NJ) and maximum likelihood (ML) methods implemented in MEGA 6 (Tamura et al. 2013). The p-distance was used in the NJ analysis. Prior to the ML phylogenetic estimation, the best nucleotide substitution model was evaluated using MEGA 6, and the GTR + G + I model was selected based on corrected Akaike information criterion (AICc) scores. Detailed information about the samples is provided in ESM Table S1. The robustness of the nodes on each tree was assessed by generating 1000 bootstrap replicates. For phylogenetic analysis, the following sequences of the same or related species were used as outgroups: Chinese “O. platypus” (accession numbers and sample ID: AY245029, XIA39-1; AY245067, CHI53-1; AY245062, LI20-1; AY245057, YU25-1; Perdices et al. 2004; KP101036, NRMT3590; KP101038, NRMT3095; KP101019, NRMT3531; KP101023, NRMT3216; KP101025, NRMT3714; KP101029, NRMT3154; KP101032, NRMT3674; KP101044, NRMT3029; KP101045, NRMT3030; KP101058, NRMT3996; KP101062, NRMT3421, KP101069, NRMT3921; Perdices and Coelho 2006), O. uncirostris uncirostris (collected for this study from Anegawa River, Nagahama, Shiga; accession number: LC021312), Candida sieboldii (from Usogawa River, Hikone, Shiga; LC021311), C. temminckii (from Adogawa River, Takashima, Shiga; LC021310), and Aphyocypris chinensis (AB218688; Saitoh et al. 2006), referring to Saitoh et al. (2011) and Hosoya (2013) . The lack of the nucleotides in some cited data were treated as missing data.
A minimum spanning network was constructed using Arlequin ver. 3.5 (Excoffier and Lischer 2010) and Hapstar (Teacher and Griffiths 2011) to assess the phylogenetic relationships among the observed haplotypes. Haplotype and nucleotide diversities at each sampling site were calculated using Arlequin.
The divergence times of each major clade of O. platypus were estimated by a Bayesian estimation method implemented in BEAST ver. 1.8.1 (Drummond and Rambaut 2007). In this analysis, the same dataset was used for the NJ and ML analyses except for the sequences in Perdices and Coelho (2006) due to their short length (940 bp). A commonly used mutation rate for cyprinid fish (i.e., 0.76 %/Myr/lineage; Zardoya and Doadrio 1999) and TN93 + G model selected by the Bayesian information criterion (BIC) in MEGA were adopted. The Yule process was used as the tree prior and we adopted the uncorrelated lognormal relaxed clock. Markov-chain Monte Carlo (MCMC) simulations were run for 40 million generations, sampling every 1,000 generations with the first 10 % of generations discarded as burn-in. We performed two independent runs and the results of these runs were combined using LogCombiner ver. 1.8.1, and the summary statistics of the estimated parameters were visualized using Tracer ver. 1.6 (Rambaut et al. 2014)
Demographic and genetic structure analyses. We conducted mismatch distribution tests for demographic expansion (Rogers and Harpending 1992) in three of the clades identified in the phylogenetic and network analyses (i.e., EJ, WJ, and KY; see “Results”). To test whether the sequence data deviated significantly from expectations under a sudden expansion model, goodness of fit based on the sum of square deviations (SSD) was calculated with 10,000 replicates. In addition, we also calculated Tajima’s D (Tajima 1989) and Fu’s Fs (Fu 1997) for these groups to infer potential expansion. All the demographic tests were performed using Arlequin. When expansions were detected, the time of sudden expansion (t) was estimated as t = τ/2u, where τ is an expansion parameter. We calculated u according to the equation u = 2µL, where µ is the molecular divergence rate per nucleotide and L is the DNA length analyzed (Rogers and Harpending 1992; Schenekar and Weiss 2011). In this study, the value used was µ = 0.76 %/Myr/lineage (Zardoya and Doadrio 1999). For WJ clade, to avoid misleading the result of the above demographic analyses (reviewed by Grant 2015), we conducted further estimation of the indices that excluded the regional subclade found in northern Kyushu (see “Results”).
In eastern to central Honshu Island, we calculated genetic differentiation, estimated by F ST, between eastern (from Ibaraki to Nagano and Shizuoka Prefectures, locality ID 3-8, 16-23, 43-46) and western part (Aichi, Gifu, and Mie Prefectures, locality ID 24-36, 38, 47-53) of the region, because there was a possibility of regional differentiation within clade (see “Results”). The significance level of F ST was obtained from 10,000 permutations using Arlequin.
Landscape-shape interpolation analysis was performed to visualize the spatial patterns of genetic diversity across the study area using Alleles In Space (Miller 2005). In this analysis, higher genetic diversity could also be found in zones where subpopulations from different geographical origins secondarily contacted or where native and introduced populations had become mixed (Miller et al. 2006). To avoid misleading results from these types of mixing, we used only samples that were thought to be from indigenous populations and conducted the analyses for each clade. In this procedure, although there are some ambiguities of detailed natural distribution, we regarded a population as an indigenous population according to Mizuguchi (1990) and excluded the WJ haplotypes in the distribution areas of EJ and KY clades (see “Results”).
Results
Phylogenetic analysis and divergence time. A total of 195 Opsariichthys platypus haplotypes were identified among 789 individuals collected from 124 localities (ESM Table S1) based on 1004 bp nucleotide sequences. These new haplotypes of cytochrome b were deposited in the DNA databank DDBJ/EMBL/GenBank (accession numbers: LC19793–LC19987; details are shown in ESM Table S2) and in a database of genetic diversity for Japanese freshwater fishes (GEDIMAP, Watanabe et al. 2010a) (data ID: P1847–1969, 1979). The haplotype and nucleotide diversities of all samples were 0.985 and 0.01665, respectively (ESM Table S1).
The NJ phylogenetic tree revealed three major O. platypus clades with high bootstrap support: western Japan (WJ), eastern Japan (EJ), and Kyushu (KY) (Fig. 2). The topology of the ML tree (not shown) were similar to the NJ trees, that is, clade KY is primarily divided from the haplotypes of Honshu, and then the Honshu haplotypes are further discriminated into two clades (EJ and WJ). Clades EJ and KY clearly correspond to a distribution area, that is, the haplotypes of EJ and KY were distributed, respectively, in eastern Honshu (from Kanto to Tokai regions) and Kyushu (except for Oita Prefecture). In contrast, the haplotypes of WJ were widely distributed throughout Japan (Fig. 3). Among the three clades, the KY showed remarkable differentiation from the other two clades and uncorrected p-distance between EJ and KY and between WJ and KY were 3.4 % and 3.0 %, respectively, while that of between WJ and EJ was 1.9 %. The haplotype and nucleotide diversities of each clade were 0.9272–0.9748 and 0.00391–0.00632, respectively (Table 1). The mean proportion of different sites (p-distance) within clades, between clades, and net between clades were 0.005–0.007, 0.019–0.034, and 0.012–0.029, respectively (ESM Table S3).

Neighbor-joining (NJ) tree of Opsariichthys platypus and outgroups based on mitochondrial cytochrome b (cyt b) sequences (1004 bp). Sixteen sequences were based on the cyt b sequences of O. platypus in China (Perdices et al. 2004; Perdices and Coelho 2006). Numbers on major nodes represent NJ/maximum likelihood (ML) bootstrap support (1000 replicates). Western Japan (WJ), eastern Japan (EJ), and Kyushu (KY) clades are colored blue, green, and red, respectively. Light-blue and yellow-green represent the Kyushu subclade of WJ and the haplotypes distributed in the western region of the EJ clade, respectively

Distribution of each clade of Opsariichthys platypus. Haplotype colors as in Fig. 2
The estimation of divergence time revealed that the differentiation between KY and the two other clades (i.e., WJ and EJ) might have occurred 1.9–4.1 million years ago (Mya) in the late Pliocene–Pleistocene. The divergence time between WJ and EJ was estimated at 1.5–3.1 Mya.
The genetic differences among Chinese O. platypus haplotypes were extensive and the haplotypes were divided into two clades (north and south China; Fig. 2). The north China clade grouped with the Japanese clades, and these clades formed a monophyletic group. However, the haplotypes of south China comprised a well-supported single clade, including O. uncirostris uncirostris.
The minimum spanning network strongly supported the results from the phylogenetic trees, indicating the presence of regional genetic structure (Fig. 4). The three major clades were separated from each other and each of them showed a bush-like pattern. The central (e.g., WJ08, WJ47, KY03, KY06) and more connected haplotypes (e.g., WJ03, EJ04) were widely distributed within each distribution area (except for WJ88, EJ08, EJ22; ESM Table S1). In the WJ clade, nearly all the WJ haplotypes in the areas thought as non-indigenous regions were the same as or similar to the haplotypes in Lake Biwa (ESM Table S1). In addition, the eight haplotypes (WJ74-81) distributed only within northern Kyushu (locality ID 89, 91-97) and Yamaguchi (locality ID 76) constituted a monophyletic subclade (Fig. 4). In the EJ clade, most of the haplotypes distributed in the eastern region (Ibaraki to Nagano and Shizuoka Prefectures) constituted a monophyletic subclade that was derived from those in the western region (Aichi, Gifu, and Mie Prefectures) (Fig. 4). The haplotype differences demonstrated genetic differentiation between the eastern and western regions in the EJ clade (F ST = 0.364, P < 0.001).

Minimum spanning network of Opsariichthys platypus derived from cytochrome b sequences. Haplotype colors as in Fig. 2
Demography and genetic structuring. A mismatch distribution test for each of the three clades did not differ significantly from the null hypothesis of population expansion and suggests the occurrence of population expansion in all of the clades (WJ: τ = 6.887, SSD = 0.003, P = 0.122; EJ: τ = 5.711, SSD = 0.010, P = 0.068; KY: τ = 3.672, SSD = 0.004, P = 0.666; Fig. 5). Similarly, Tajima’s D and Fu’s Fs for the three clades exhibited significant negative values (except for Tajima’s D in clade KY), indicating the possibility of the occurrence of population expansion (Table 1). The estimated times since the expansion of WJ, EJ, and KY were approximately 226,000, 187,000, and 120,000 years, respectively. In further analyses of clade WJ without regional subclade, the occurrence of population expansion was indicated (τ = 6.594, SSD = 0.001, P = 0.460). Tajima’s D and Fu’s Fs also indicated a similar trend (D = -1.92, P = 0.002; Fs = -24.60, P = 0.001) and the time since expansion was approximately 216,000.

Mismatch distribution based on cytochrome b sequences for the three clades of Opsariichthys platypus. Solid lines and dotted lines represent the expected distribution under the sudden expansion model and the 95 % confidence interval, respectively. Open circles represent the observed frequencies of pairwise differences
Landscape-shape interpolation analysis revealed the geographic locations of higher genetic diversity in each clade (Fig. 6). In this analysis, the populations that were thought to be artificially introduced in accordance to Mizuguchi (1990) were excluded. Although WJ haplotypes were widely distributed, those in eastern-central Honshu and in southwestern Kyushu were also excluded because those haplotypes are non-indigenous (see “Discussion”). In the WJ clade, geographic regions with higher genetic diversities were found in the Kinki district, and the lowest genetic diversity was found in the northern Kyushu district except around Kanmon Strait. In the EJ clade, populations from the central region showed higher genetic diversities than those from northern Tokai, and the lowest genetic diversity was observed in the Kanto district. In the KY clade, populations from the southern region showed higher genetic diversity (Fig. 6).

Results of a landscape-shape interpolation analysis of each mitochondrial DNA clade. The geographic regions with the highest genetic diversity are shown in red, and blue represents the areas of lowest genetic diversity
Discussion
The present study revealed that the mtDNA lineage of Japanese pale chub Opsariichthys platypus is divided into three major clades (western Japan: WJ; eastern Japan: EJ; and Kyushu: KY) (Figs. 2, 4), and the distribution of the EJ and KY clades clearly reflects their respective geographical ranges (Fig. 3). These results suggest the existence of regional differentiation in O. platypus. On the other hand, clade WJ is distributed throughout the Japanese Archipelago, including the eastern and northern part of Honshu (Tohoku, Hokuriku, and San-in districts), western Shikoku, southern Kyushu, and Tokunoshima Island, where O. platypus was not distributed naturally (Fig. 3; Mizuguchi 1990). The presence of O. platypus in non-indigenous regions had been suggested to be the result of its inadvertent introduction from Lake Biwa in conjunction with the Ayu Plecoglossus altivelis altivelis, one of the most important fish for freshwater fisheries in Japan (Nakamura 1969; Mizuguchi 1990; Ma et al. 2006; Takamura and Nakahara 2015). In this study, nearly all the WJ haplotypes in the non-indigenous regions were the same as or similar to the haplotypes in Lake Biwa (ESM Table S1). The WJ haplotypes in the distribution ranges of the EJ and KY clades were also same as or similar to those of Lake Biwa; thus, the WJ haplotypes may have been introduced into various regions by artificial transplantation. However, the WJ haplotypes in southern Kyushu and Tokunoshima were the same as those in Oita Prefecture, northeastern Kyushu (ESM Table S1). This indicates that the WJ haplotypes in northern Kyushu were transplanted into that area. In addition, the EJ and KY clades might be also introduced to non-indigenous regions that were adjacent to their original distribution ranges (Fig. 3). Mizuguchi (1990) reported that the population of O. platypus on the Izu Peninsula, Shizuoka Prefecture, had originated by transplantation from the Tenryu River, Shizuoka Prefecture. Evidence of transplantation of O. platypus among the rivers in Kyushu was obtained by hearing to freshwater fisheries cooperatives in Kyushu (Onikura, unpublished). Thus, the geographical distributions of the EJ and KY clades may also be somewhat disturbed by artificial transplantation.
Among the three clades of O. platypus, the KY clade showed remarkable differentiation from the other two clades. Clade KY is only distributed in Kyushu, except for Oita Prefecture, and this suggests that the mtDNA lineage of O. platypus is divided by the Kanmon Strait (between Honshu and Kyushu) and the eastern part of the Chikushi Mountains (between Oita Prefecture and the rest of the northern Kyushu region). Such mtDNA divergence in northern Kyushu had been reported in some freshwater fishes, including Oryzias latipes (Takehana et al. 2003), Tanakia limbata (Hashiguchi et al. 2006; Matsuba et al. 2014), Rhodeus atremius (Miyake et al. 2011), Hemigrammocypris rasborella (Watanabe et al. 2014), and Pseudogobio esocinus (Tominaga et al. 2016). A parsimony analysis of endemicity (PAE) of the Japanese primary freshwater fish fauna based on the literature (Watanabe 1998, 2012) and a clustering analysis of the northern Kyushu freshwater fish fauna based on field surveys (Nakajima et al. 2006) also indicated that the northern Kyushu freshwater fish fauna is divided by the eastern part of Chikushi Mountains (Sangun Mountains), so that the fish fauna in northeastern Kyushu facing the Seto Inland Sea (i.e., the eastern side of the Sangun Mountains and Oita Prefecture) is similar to that of Honshu and Shikoku rather than to the other regions within Kyushu. For example, a striped loach Cobitis biwae that is distributed in western Honshu is also distributed in Oita Prefecture, but it is not found in the other regions of Kyushu (Kitagawa et al. 2004). In the present study, the KY clade of O. platypus was absent in this prefecture, while the WJ clade was present. These biogeographic concordances suggest that the eastern part of the Chikushi Mountains separates the freshwater fish populations.
The second largest differentiation was found between the WJ and EJ clades, and the boundary geographically separating these clades seem to correspond with the Ibuki–Suzuka Mountains. Many studies have reported a genetic border in freshwater fish populations at the Ibuki–Suzuka Mountains (Matsuda et al. 1997; Saka et al. 2003; Takehana et al. 2003; Watanabe and Mori 2008; Tominaga et al. 2009, 2016; Miyazaki et al. 2011; Komiya et al. 2014; Watanabe et al. 2014). Because the time of uplifting of the Ibuki–Suzuka Mountains (ca. 1.0–1.5 Mya; Kawabe 1994; Kaizuka et al. 2000) is consistent with the estimated divergence time between the WJ and EJ clades of O. platypus (ca. 1.5–3.1 Mya) and those of other freshwater fishes to some extent, long-term separation is a plausible explanation for the genetic differentiation between the western and eastern populations of O. platypus.
On the other hand, the Fossa Magna in central Honshu Island is regarded as the most important factor in structuring the Japanese freshwater fish fauna, as reported in other studies (e.g., Watanabe et al. 2000; Kitagawa et al. 2003; Saka et al. 2003; Mabuchi et al. 2008; Miyazaki et al. 2011; Tominaga et al. 2016; reviewed by Watanabe et al. 2006 and Watanabe 2012). Although some freshwater fishes have large genetic divergences between the eastern and western regions of the Fossa Magna, the mtDNA phylogeny of the O. platypus was not divided at this point. A similar pattern had already reported in Oryzias latipes (Takehana et al. 2003), so there is a possibility that some freshwater fishes could have expanded their distribution beyond the Fossa Magna region. However, in the EJ clade of O. platypus, the eastern (EJ13, EJ15–33, EJ46–48) and western haplotypes (EJ01–12, EJ14, EJ34–45, EJ49–50) were not shared between the two regions and the haplotype differences clearly showed genetic differentiation between the eastern and western populations in the EJ clade (F ST = 0.364, P < 0.001). These results indicate that O. platypus have a certain level of genetic divergences between the eastern and western regions of the Fossa Magna, even though the haplotypes in each region did not make monophyletic clades (Fig. 4). In addition, if two haplotypes (EJ13 and EJ48) were artificially introduced from Tokai to Kanto district, the eastern and western populations of EJ clade were more clearly separated. However, further studies are needed to identify indigenous haplotypes in local regions.
In the WJ clade, some geographical differentiation was also observed. Although most of the WJ haplotypes were same or close to those in Lake Biwa, eight haplotypes (WJ74–81) in northern Kyushu and Yamaguchi constituted a monophyletic subclade (Fig. 4; Table S1). This result suggests that the subclade had been originally distributed in northern Kyushu and adjacent areas of Honshu, and it became isolated from the other WJ haplotypes. Studies of other freshwater fishes such as Pseudogobio esocinus and Oryzias latipes found a similar geographical distribution pattern of local mitochondrial lineages in the area (Takehana et al. 2003; Tominaga et al. 2016). Thus, several freshwater fish populations in the northern Kyushu–Yamaguchi area might have been isolated from those in other Honshu and Kyushu areas.
In Kyushu, although the KY clade did not show any regional geographical differentiation, the genetic landscape-shape interpolation analysis showed higher genetic diversity in the southern area (Fig. 6). This result indicates that the distribution of the KY clade might have expanded from south to north. In the northernmost area of Kyushu, however, the northern Kyushu subclade of WJ haplotypes was distributed with the KY clade. This overlapping might have resulted from secondary contact between the two lineages. The northern Kyushu subclade of WJ consisted of endemic monophyletic haplotypes, and thus it had been distributed in the area for a long time after the population expansion of the WJ clade (approximately, 226,000 years ago as estimated by mismatch distribution analysis; Fig. 5). In contrast, the KY clade in the area showed lower genetic diversity, indicating recent immigration. These results suggest that the northern Kyushu subclade of WJ had been established earlier than the northward expansion of the KY clade. In this study, however, we could not determine whether the invasion of the KY clade into northernmost Kyushu was a prehistoric or an anthropogenic event.
In the present study, we found that O. platypus has clear genetic differentiation among regional populations, and this emphasizes the need to protect such indigenous genetic features as important conservation units (Frankham et al. 2004). The fact that haplotypes of WJ were introduced throughout the Japanese Archipelago could indicate an ongoing loss of the genetic features of the Kyushu and eastern Honshu populations due to introductions or introgression from western Honshu populations. Takamura and Nakahara (2015) indicated that populations of O. platypus in major rivers in the Kanto Plain had already been admixed with introduced populations from Lake Biwa. Therefore, efforts should be made toward a comprehensive understanding of the current state of introductions of non-indigenous O. platypus populations and the potential influence of invasive populations, including loss of genetic diversity.
References
Biodiversity Center of Japan, Nature Conservation Bureau, Ministry of the Environment (2010) Japan Wildlife Research Center (ed) Biodiversity of Japan; a harmonious coexistence between nature and humankind. Heibonsha, Tokyo
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Frankham R, Ballow JD, Briscoe DA (2004) A primer of conservation genetics. Cambridge University Press, Cambridge
Fu Y-X (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Grant WS (2015) Problems and cautions with sequence mismatch analysis and Byesian skyline plots to infer historical demography. J Hered 106:33–346
Hashiguchi Y, Kado T, Kimura S, Tachida H (2006) Comparative phylogeography of two bitterlings, Tanakia lanceolata and T. limbata (Teleostei, Cyprinidae), in Kyushu and adjacent districts of western Japan, based on mitochondrial DNA analysis. Zool Sci 23:309–322
Honda M, Matsui M, Tominaga A, Ota H, Tanaka S (2012) Phylogeny and biogeography of the Anderson’s crocodile newt, Echinotriton andersoni (Amphibia: Caudata), as revealed by mitochondrial DNA sequences. Mol Phylogenet Evol 65:642–653
Hosoya K (2013) Cyprinidae. In: Nakabo T (ed) Fishes of Japan with pictorial keys to the species, third edn. Tokai University Press, Hadano, pp 308–327, 1813–1819
Kaizuka S, Koike K, Endo K, Yamazaki H, Suzuki H (2000) Regional geomorphology of the Japanese Islands, vol. 4 Geomorphology of Kanto and Izu-Ogasawara. University of Tokyo Press, Tokyo
Kakioka R, Kokita T, Tabata R, Mori S, Watanabe K (2013) The origins of limnetic forms and cryptic divergence in Gnathopogon fishes (Cyprinidae) in Japan. Environ Biol Fish 96:631–644
Kawabe T (1994) Biwako no oitachi (formation of Lake Biwa). In: Biwa Research Group for Natural History of Lake Biwa (ed) Biwako no shizenshi (The natural history of Lake Biwa). Yasaka Shobo, Tokyo, pp 24–72
Kitagawa E, Hoshino K, Okazaki T, Kitagawa T (2004) Cobitis biwae from Oita River system in Oita Prefecture, Japan, and its biogeographic origin. Jpn J Ichthyol 51:117–122
Kitagawa T, Yoshioka M, Kashiwagi M, Okazaki T (2001) Population structure and local differentiation in the delicate loach, Niwaella delicata, as revealed by mitochondrial DNA and morphological analyses. Ichthyol Res 48:127–135
Kitagawa T, Watanabe M, Kitagawa E, Yoshioka M, Kashiwagi M, Okazaki T (2003) Phylogeography and the maternal origin of the tetraploid form of the Japanese spined loach, Cobitis biwae, revealed by mitochondrial DNA analysis. Ichthyol Res 50:318–325
Koizumi I, Usio N, Kawai T, Azuma N, Masuda R (2012) Loss of genetic diversity means loss of geological information: the endangered Japanese crayfish exhibits remarkable historical footprints. PLoS One 7:e33986
Komiya T, Fujita-Yanagibayashi S, Watanabe K (2014) Multiple colonizations of Lake Biwa by Sarcocheilichthys fishes and their population history. Environ Biol Fish 97:741–755
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and clustal X ver 2.0. Bioinformatics 23:2947–2948
Ma GC, Watanabe K, Tsao HS, Yu HT (2006) Mitochondrial phylogeny reveals the artificial introduction of the pale chub Zacco platypus (Cyprinidae) in Taiwan. Ichthyol Res 53:323–329
Mabuchi K, Senou H, Nishida M (2008) Mitochondrial DNA analysis reveals cryptic large-scale invasion of non-native genotypes of common carp (Cyprinus carpio) in Japan. Mol Ecol 17:796–809
Machida H, Ota Y, Kawana T, Moriwaki H, Nagaoka S (2001) Regional geomorphology of the Japanese Islands, vol. 7, geomorphology of Kyushu and Ryukyus. University of Tokyo Press, Tokyo
Machida H, Matsuda T, Umitsu M, Koizumi T (2006) Regional geomorphology of Japanese Islands, geomorphology of Chubu, vol 5. University of Tokyo Press, Tokyo
Matsuba H, Yoshimi S, Inoue M, Hata H (2014) Origin of Tanakia limbata in Ehime Prefecture indicated by phylogeographic analysis of mitochondrial cytochrome b sequences. Jpn J Ichthyol 61:89–96
Matsuda M, Yonekawa H, Hamaguchi S, Sakaizumi M (1997) Geographic variation and diversity in the mitochondrial DNA of the medaka, Oryzias latipes, as determined by restriction endonuclease analysis. Zool Sci 14:517–526
Millien-Parra V, Jaeger J-J (1999) Island biogeography of the Japaese terrestrial mammal assemblages: an example of a relict fauna. J Biogeogr 26:959–972
Miller MP (2005) Alleles In Space (AIS): Computer software for the joint analysis of interindividual spatial and genetic information. J Hered 96:722–724
Miller MP, Renee Bellinger M, Forsman ED, Haig SM (2006) Effects of historical climate change, habitat connectivity, and vicariance on genetic structure and diversity across the range of the red tree vole (Phenacomys longicaudus) in the Pacific Northwestern United States. Mol Ecol 15:145–159
Miyake T, Nakajima J, Onikura N, Ikemoto S, Iguchi K, Komaru A, Kawamura K (2011) The genetic status of two subspecies of Rhodeus atremius, an endangered bitterling in Japan. Conserv Genet 12:282–400
Miyazaki JI, Dobashi M, Tamura T, Beppu S, Sakai T, Mihara M, Hosoya K (2011) Parallel evolution in eight-barbel loaches of the genus Lefua (Balitoridae, Cypriniformes) revealed by mitochondrial and nuclear DNA phylogenies. Mol Phylogenet Evol 60:416–427
Mizuguchi K (1990) Dispersal of the oikawa, Zacco platypus (temminck et schlegel), in Japan. Rep Tokyo Univ Fish 25:149–169
Morishima K, Nakamura-Shiokawa Y, Bando E, Li Y-J, Boron A, Khan MMR, Arai K (2008) Cryptic clonal lineage and genetic diversity in the loach Misgurnus anguillicaudatus (Teleostei: Cobitidae) inferred from nuclear and mitochondrial DNA analyses. Genetica 132:159–171
Nakagawa H, Seki S, Ishikawa T, Watanabe K (2016) Genetic population structure of the Japanese torrent catfish Liobagrus reinii (Amblycipitidae) inferred from mitochondrial cytochrome b variations. Ichthyol Res doi 10.1007/s10228-015-0503-6
Nakajima J, Onikura N, Matsui S, Oikawa S (2006) Geographical distribution of genuine freshwater fishes in Fukuoka Prefecture, northern Kyushu, Japan. Jpn J Ichthyol 53:117–131
Nakamura M (1969) Cyprinid Fishes of Japan. Research Institute for Natural Resourdes, Tokyo
Nishizawa T, Kurabayashi A, Kunihara T, Sano N, Fujii T, Sumida M (2011) Mitochondrial DNA diversification, molecular phylogeny, and biogeography of the primitive rhacophorid genus Buergeria in East Asia. Mol Phylogenet Evol 59:139–147
Nunome M, Torii H, Matsuki R, Kinoshita G, Suzuki H (2010) The Influence of Pleistocene Refugia on the Evolutionary History of the Japanese Hare, Lepus brachyurus. Zool Sci 27:746–754
Oba H (1994) The flora of Japan and the implication of global climatic change. J Plant Res 107:85–80
Ohnishi N, Uno R, Ishibashi Y, Tamate HB, Oi T (2009) The influence of climatic oscillations during the Quaternary Era on the genetic structure of Asian black bears in Japan. Heredity 102:579–589
Ota (2000) The current geographic faunal pattern of reptiles and amphibians of the Ryukyu Archipelago and adjacent regions. Tropics 10:51–62
Perdices A, Coelho MM (2006) Comparative phylogeography of Zacco platypus and Opsariichthys bidens (Teleostei, Cyprinidae) in China based on cytochrome b sequences. J Zool Syst Evol Res 44: 330–338
Perdices A, Cunha C, Coelho MM (2004) Phylogenetic structure of Zacco platypus (Teleostei, Cyprinidae) populations on the upper and middle Chang-Jiang (= Yangtze) drainage inferred from cytochrome b sequences. Mol Phylogenet Evol 31:192–203
Rambaut A, Suchard MA, Xie D, Drummond AJ. (2014) Tracer ver 1.6. Available from http://beast.bio.ed.ac.uk/Tracer
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Saitoh K, Sado T, Mayden RL, Hanzawa N, Nakamura K, Nishida M, Miya M (2006) Mitogenomic evolution and interrelationships of the cypriniformes (Actinopterygii : Ostariophysi): The first evidence toward resolution of higher-level relationships of the world’s largest freshwater fish clade based on 59 whole mitogenome sequences. J Mol Evol 63:826–841
Saitoh K, Sado T, Doosey MH, Bart Jr HL, Inoue JG, Nishida M, Mayden RL, Miya M (2011) Evidence from mitochondrial genomics supports the lower Mesozoic of South Asia as the time and place of basal divergence of cypriniform fishes (Actinopterygii: Ostariophysi). Zool J Linn Soc 161:633–662
Saka R, Takehana Y, Suguro N, Sakaizumi M (2003) Genetic population structure of Lefua echigonia inferred from allozymic and mitochondrial cytochrome b variations. Ichthyol Res 50:301–309
Sasaki T, Kartavtsev YP, Chiba SN, Uematsu T, Sviridov VV, Hanzawa N (2007) Genetic divergence and phylogenetic independence of Far Eastern species in subfamily Leuciscinae (Pisces: Cyprinidae) inferred from mitochondrial DNA analyses. Genes Genet Syst 82:329–340
Schenekar T, Weiss S (2011) High rate of calculation errors in mismatch distribution analysis results in numerous false inferences of biological importance. Heredity 107:511–512
Schoville SD, Uchifune T, Machida R (2013) Colliding fragment islands transport independent lineages of endemic rock-crawlers (Grylloblattodea: Grylloblattidae) in the Japanese archipelago. Mol Phylogenet Evol 66:915–927
Suzuki T, Kitano T, Tojo K (2014) Contrasting genetic structure of closely related giant water bugs: Phylogeography of Appasus japonicus and Appasus major (Insecta: Heteroptera, Belostomatidae). Mol Phylogenet Evol 72:7–16
Tajima F (1989) Statistical method for testing the nautral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Takamura K, Nakahara M (2015) Intraspecific invasion occurring in geographically isolated populations of the Japanese cyprinid fish Zacco platypus. Limnology doi 10.1007/s10201-015-0450-y
Takehana Y, Nagai N, Matsuda M, Tsuchiya K, Sakaizumi M (2003) Geographic variation and diversity of the cytochrome b gene in Japanese wild populations of Medaka, Oryzias latipes. Zool Sci 20:1279–1291
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol 30:2725–2729
Teacher AGF, Griffiths DJ (2011) HapStar: automated haplotype network layout and visualization. Mol Ecol Resour 11:151–153
Tominaga K, Watanabe K, Kakioka R, Mori S, Jeon SR (2009) Two highly divergent mitochondrial DNA lineages within Pseudogobio esocinus populations in central Honshu, Japan. Ichthyol Res 56:195–199
Tominaga A, Matsui M, Yoshikawa N, Nishikawa K, Hayashi T, Misawa Y, Tanabe S, Ota H (2013) Phylogeny and historical demography of Cynops pyrrhogaster (Amphibia: Urodela): Taxonomic relationships and distributional changes associated with climatic oscillations. Mol Phylogenet Evol 66:654–667
Tominaga K, Nakajima J, Watanabe K (2016) Cryptic divergence and phylogeography of the pike gudgeon Pseudogobio esocinus (Teleostei: Cyprinidae): a comprehensive case of freshwater phylogeography in Japan. Ichthyol Res 63:79–93
Watanabe K (1998) Parsimony analysis of the distribution pattern of Japanese primary freshwater fishes, and its application to the distribution of the bagrid catfishes. Ichthyol Res 45:259–270
Watanabe K (2012) Faunal structure of Japanese freshwater fishes and its artificial disturbance. Environ Biol Fishes 94:533–547
Watanabe K, Mori S (2008) Comparison of genetic population structure between two cyprinids, Hemigrammocypris rasborella and Pseudorasbora pumila subsp., in the Ise Bay basin, central Honshu, Japan. Ichthyol Res 55:309–320
Watanabe K, Iguchi K, Hosoya K, Nishida M (2000) Phylogenetic relationships of the Japanese minnows, Pseudorasbora (Cyprinidae), as inferred from mitochondrial 16S rRNA gene sequences. Ichthyol Res 47:43–50
Watanabe K, Takahashi H, Kitamura A, Yokoyama R, Kitagawa T, Takeshima H, Sato S, Yamamoto S, Takehana Y, Mukai T, Ohara K, Iguchi K (2006) Biogeographical history of Japanese freshwater fishes: Phylogeographic approaches and perspectives. Jpn J Ichthyol 53:1–38
Watanabe K, Kano Y, Takahashi H, Mukai T, Kakioka R, Tominaga K (2010a) GEDIMAP: a database of genetic diversity for Japanese freshwater fishes. Ichthyol Res 57:107-109
Watanabe K, Kawase S, Mukai T, Kakioka R, Miyazaki J, Hosoya K (2010b) Population divergence of Biwia zezera (Cyprinidae: Gobioninae) and the discovery of a cryptic secies, based on mitochondrial and nuclear DNA sequence analyses. Zool Sci 27:647–655
Watanabe K, Mori S, Tanaka T, Kanagawa N, Itai T, Kitamura J, Suzuki N, Tominaga K, Kakioka R, Tabata R, Abe T, Tashiro Y, Hashimoto Y, Nakajima J, Onikura N (2014) Genetic population structure of Hemigrammocypris rasborella (Cyprinidae) inferred from mtDNA sequences. Ichthyol Res 61:352–360
Yamazaki Y, Goto A, Nishida M (2003) Mitochondrial DNA sequence divergence between two cryptic species of Lethenteron, with referene to an improved identification technique. J Fish Biol 62:591–609
Yonekura N, Kaizuka S, Nogami M, Chinzai K (2001) Regional geomorphology of the Japanese Islands, vol 1. Introduction to Japanese geomorphology. University of Tokyo Press, Tokyo
Zardoya R, Doadrio I (1999) Molecular evidence on the evolutionary and biogeographical patterns of European cyprinids. J Mol Evol 49:227–237
Acknowledgments
We are grateful to S. Kitano (Nagano Nature Conservation Research Institute), J. Mima (Environmental Assessment Center CO., LTD), T. Yodo (Mie University), K. Hirashima (Wakayama Prefectural Museum of Natural History), T. Tsuruta (Osaka Sangyo University), M. Shibuya (Sumiko Techno Research, Co., Ltd), T. Nomura (Kanagawa Prefecture), T. Kitamura (Akankohan Eco Museum Center), T. Shimizu (Ehime Prefectural Chuyo Fisheries Experimental Station), T. Ohnaka (Aichi Prefecture), T. Abe (Okayama University), K. Sakai (Noto Marine Center), and Y. Koya, C. Sato, Y. Takeuchi, Y. Ono, A. Tanaka, C. Kato, R. Sakai (Gifu University) for their cooperation with the collection of specimens and laboratory experiments. Hiroshi Takahashi and the two anonymous reviewers also provided comments that helped to improve the manuscript. This work was supported by the Global Environment Research Fund (RF-075 and RF-0910) of the Ministry of the Environment, Japan, and by JSPS KAKENHI Grant Numbers 21370035 and 26250044.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the Electronic Supplementary Material.
About this article

Cite this article
Kitanishi, S., Hayakawa, A., Takamura, K. et al. Phylogeography of Opsariichthys platypus in Japan based on mitochondrial DNA sequences. Ichthyol Res 63, 506–518 (2016). https://doi.org/10.1007/s10228-016-0522-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10228-016-0522-y