
Abstract
Background
Autosomal dominant polycystic kidney disease (ADPKD), one of the most common hereditary kidney diseases, causes gradual growth of cysts in the kidneys, leading to renal failure. Owing to the advanced technology of next-generation sequencing (NGS), genetic analyses of the causative genes PKD1 and PKD2 have been improved.
Methods
We performed genetic analyses of 111 Japanese ADPKD patients using hybridization-based NGS and long-range (LR)-PCR-based NGS. Additionally, genetic analyses in exon 1 of PKD1 using Sanger sequencing because of an extremely low coverage of NGS and those using multiplex ligation-dependent probe amplification (MLPA) were performed.
Results
The detection rate using NGS for 111 patients was 86.5%. One mutation in exon 1 of PKD1 and five deletions detected by MLPA were identified. When combined, the total detection rate was 91.9%.
Conclusion
Although NGS is useful, we propose the addition of Sanger sequencing for exon 1 of PKD1 and MLPA as indispensable for identifying mutations not detected by NGS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD), one of the most common hereditary kidney diseases, causes gradual growth of cysts in the kidneys, leading to renal failure. Two causative genes were PKD1 and PKD2. Polycystin 1 (PC1), encoded by PKD1, assists in mechanosensation of urine flow in the cilia of renal tubules. Polycystin 2 (PC2), encoded by PKD2, functions as a calcium channel in cooperation with PC1. PC1 and PC2 play a critical role in suppression of renal tubule dilation and the dysfunction of either gene may lead to renal tubule expansion and cyst formation [1].
The average age at onset of ESRD is 58 years for those with PKD1 mutations and 79 years for those with PKD2 mutations. In patients with PKD1 mutations, the age of ESRD is 55 years for those with a truncating mutation, while it is 67 years for those with a non-truncating mutation [2]. The effectiveness of tolvaptan, a vasopressin receptor antagonist, in treating ADPKD was reported in 2012 [3]. According to a sub-analysis of this clinical trial, early treatment in patients with a truncating mutation in PKD1 was desirable when compared with that in patients with mutations in PKD2 or a non-truncating mutation in PKD1 [4]. The search for genetic mutations has been useful for adapting such drugs, and demand for genetic analyses may increase in future.
PKD1 and PKD2 are large genes comprising 46 exons and 15 exons, respectively. Approximately, two-thirds of the 5′ region of PKD1 constitute a region called the “duplicated region”. This region is 95–98% homologous to six pseudogenes within chromosome 16 (16p13.1). Therefore, for mutation analyses in this region, long-range PCR (LR-PCR) that only amplifies genuine PKD1 using primer sets specific PKD1 sequences, followed by direct sequencing, has been established [5,6,7]. These analyses are very complex and, as such, take a significant amount of time for completion.
The recent development of next-generation sequencing (NGS) has brought about changes in genetic analysis methods in ADPKD. NGS analyses using a LR-PCR-based target enrichment method (LR-PCR-based NGS) and those using a hybridization-based target enrichment method (hybridization-based NGS) have both genetic mutation detection rates of 70% [8, 9]. However, when combined with recent technological improvements in sequencers, a LR-PCR-based NGS has a detection rate of 89% [10]. NGS has reduced the complexity and turnaround time compared with previous genetic analyses. The multiplex ligation-dependent probe amplification (MLPA) method, which detects large rearrangements, is also a useful genetic analysis method for diagnosis of ADPKD [11, 12].
In this study, we performed genetic analyses of Japanese patients with ADPKD using both hybridization-based and LR-PCR-based NGS. In addition, MLPA proved beneficial for the purpose of identifying previously undetected mutations.
Materials and methods
1. Human subjects and DNA samples
We recruited 111 patients who visited Tokyo Women’s Medical University from 2010 to 2016 and who were diagnosed with ADPKD using previously established diagnostic criteria [13]. Atypical patients based on renal imaging for ADPKD were excluded [14]. Genomic DNA was extracted from peripheral blood lymphocytes using QIAamp DNA Blood Maxi Kit (QIAGEN Inc.).
2. NGS using a hybridization-based target enrichment method
Target DNA enrichment of PKD1 and PKD2 were performed using SureSelect Target Enrichment System design (Agilent, Santa Clara, CA). The design contained 120-mer baits spanning the entire non-repetitive sequence of both genes, including all exons, introns, 10 bases from the 3′ end and 10 bases from the 5′ end of each gene. Genomic coordinates of the two genes were determined using the February 2009 build (NCBI37/hg19) of the human genome in the Ensemble genome browser 12. The density of bait tiling was fivefold, and baits were allowed to overlap into repeat regions by 30 bp. The total targeted DNA length was 117 kb. All libraries were generated from sheared DNA (Covaris, Woburn, MA) with an average insert size of 200 bp following reactions with the SureSelect Target Enrichment System XT (Agilent). The enriched libraries were quantified with Bioanalyzer (Agilent). The quantified libraries were sequenced using the SOLiD 4 system (Applied Biosystems) by the paired end reading method according to the manufacture’s instruction. Sequencing data were processed with Bioscope (Applied Biosystems) and analysed with an in-house analytical pipeline. Bait settings were performed using both cDNA and genomic DNA sequencing that targeted PKD1 and PKD2 exon regions.
3. LR-PCR for NGS
To cover all exon regions of PKD1 and PKD2, PCR primers were set for 11 LR-PCRs (Fig. 1) [5, 8]. By separating LR-PCR into four groups (C18, F16, G22, and G25), multiple gene regions were amplified (Fig. 1). LR-PCR reactions with specific primers (Supplementary Table 1) were performed using KOD FX Neo (TOYOBO Inc.) as follows: for LR-PCR of C18, F16: and PKD1-1, 94 °C for 2 min, 40 cycles of 98 °C for 10 s, 68 °C for 3 min, and final extension at 72 °C for 5 min and for LR-PCR of G22, G25, and PKD2 3–7: 94 °C for 2 min, 40 cycles of 98 °C for 10 s, 68 °C for 17 min, and final extension at 72 °C for 5 min.

The gene map and position of long-range PCR (LR-PCR) products of PKD1 and PKD2. a LR-PCR products and the PKD1 pseudogenes (P1–P6) are shown below the exon–intron structure of PKD1. b LR-PCR products are shown below the exon–intron structure of PKD2
4. NGS using a LR-PCR-based target enrichment method
The LR-PCR products were purified using Agencourt AMPure XP (Beckman Coulter Inc.). The purified products were constructed into sequencing libraries using the Ion Xpress Plus Fragment Library Kit (Thermo Fisher Scientific Inc.) by means of random shearing, end-polished, and adaptor ligation. The constructed libraries were quantified using Bioanalyzer system (Agilent). The quantified libraries were used for preparation of sphere particles with Ion One Touch™ 2 (Thermo Fisher Scientific) and subsequently sequenced using Ion PGM™ sequencer (Thermo Fisher Scientific). Sequencing data were processed with Torrent Suit software (Thermo Fisher Scientific). All procedures were performed according to the manufacturer’s instructions.
5. Sanger sequencing to confirm the above-referenced NGS-detected mutations
Sanger sequencing was performed to confirm the above-referenced NGS-detected mutations. Specific primers from LR-PCR products were used for nested PCR on exons 2–34 in the PKD1 duplicated region (Supplementary Table 2) [5,6,7]. Exons 35–46 in the PKD1 single copy region and PKD2 exons were amplified using the aforementioned primers [15, 16]. The PCR products were sequenced using the BigDye Terminator v1.1 Cycle Sequencing Kit (Thermo Fisher Scientific Inc.), and evaluated using a 3130xl Genetic Analyzer (Thermo Fisher Scientific Inc.). The sequence, hg18 (PKD1: NG_008617, PKD2: NG_008604) was used as a reference sequence to compare the analysed sequences. (https://www.ncbi.nlm.nih.gov/nuccore/NG_008617, https://www.ncbi.nlm.nih.gov/nuccore/NG_008604).
6. Sanger sequencing for exon 1 in PKD1
Sanger sequencing for exon 1 in PKD1 was performed on patients whose mutations were not identified using NGS. Specific primers with LR-PCR products were used for nested PCR on exon 1 in PKD1 (Supplementary Table 2). The sequencing procedure was same as above.
7. Multiplex ligation-dependent probe amplification (MLPA)
MLPA analysis was performed on patients whose mutations were not identified using NGS [11]. The SALSA MLPA PKD1 (P351) and PKD2 (P352) kits were purchased from MRC-Holland, Inc. (Amsterdam, Netherlands), and analysis was performed using the manufacturer’s protocols.
8. Novel substitution evaluation
Analyses were performed using PKD Target (World Fusion, Tokyo, Japan) that were evaluated as previously described [10]. Except for the previously identified mutations in the PKD mutation database (PKDB) (http://pkdb.mayo.edu), we evaluated the novel mutations according to total score as described in a previous report [17]. Total score was calculated based on pathogenicity mutation determination result in the Grantham matrix scoring system [18], Align Grantham Variation Grantham Deviation (A-GVGD) [19], PolyPhen-2 [20], Sorting Intolerant from Tolerant (SIFT) [21, 22], and Mutation Taster [23].
The score was given with the following rule: a Grantham matrix score < 60 corresponded to a score of 0, and a Grantham matrix score ≥ 60 corresponded to a score of 2. An A-GVGD score from C0 to C65 corresponded to a score of from 0 to 6, respectively. For PolyPhen-2, benign, possibly damaging, and probably damaging corresponded to a score of 0, 2, and 4, respectively. For SIFT, tolerated and damaging corresponded to a score of 0 and 4, respectively. For a Mutation Taster, polymorphism and disease causing corresponded to a score of 0 and 4, respectively. A novel substitution with a total score ≥ 14 was included as a pathogenic mutation. Furthermore, the novel substitution with a combined score < 14, but with the University of California, Santa Cruz (UCSC) Vertebrate Conservation Score (GATK Guide article. Adding genome annotations using SnpEff VariantAnnotator. Available: http://www.broadinstitute.org/gatk/guide/) indicating “1-Likely pathogenic”, was also included as a pathogenic mutation.
Finally, we checked the minor allele frequencies by 1000 genomes [24] and Human Genetic Variant Database (HGVD) [25].
9. Identification of large deletion regions
PCR was performed using new primers created for the identification of regions with large deletions discovered using LR-PCR and MLPA (Supplementary Table 3).
Results
NGS using a hybridization-based targeted enrichment method
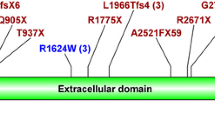
Firstly, a hybridization-based NGS was performed on 96 patients. Among these, mutations were identified in 76 patients (mutation detection rate, 79.2%). Most exons individually displayed sufficient coverage, but exons 1 and 42 of PKD1 and in exon 1 of PKD2 had low coverage sections. When breaking down the variants of 76 patients with identified mutations, 15 frameshift mutations due to insertions or deletions, 30 nonsense mutations, five splicing mutations, one in-frame deletion, and 25 substitutions were identified (Fig. 2, Supplementary Table 4). Separating the genes, 63 (82.9%) patients had PKD1 mutations and 13 (17.1%) had PKD2 mutations (Table 1). By mutation type, PKD1 mutations comprised a large number of substitutions (36.5%) and nonsense mutations (34.9%), whereas PKD2 mutations were largely nonsense mutations (61.5%) and few substitutions (15.4%) (Table 1).

Summary of mutation analyses in ADPKD
NGS using a LR-PCR-based targeted enrichment method
Multiplex LR-PCR were used groups C13, F3, G6, and PKD2 exons 3–7 (Supplementary Fig. 1). Initially, we performed genetic analysis on 19 patients whose mutations were not identified using the above-mentioned hybridization-based NGS. The coverage of PKD1 exons 1 and 42 and PKD2 exon 1 was low, similar to hybridization-based NGS.
When breaking down the variants of nine patients with identified mutations, we observed one large deletion and seven frameshift mutations in PKD1, whereas one nonsense mutation was observed in PKD2 (Fig. 2, Table 1, Supplementary Table 5). The large deletion was identified during LR-PCR within PKD1 exons 22–34. A normal 10,524-bp band and a shorter band by approximately 2 kb were detected in one patient (Fig. 3a). As a result of NGS, the coverage of exons 27–30 was less than half of that of other patients, leading us to suspect a deletion within that region (Fig. 3b). Thus, primers IF271 and IR301 were set in IVS26 and IVS30, respectively. A small band (approximately 8 kb) of the LR-PCR product from exons 22–34 was purified and used as a template for nested PCR with IF271 and IR301. This PCR product (approximately 850 bp) was sequenced (Fig. 3c, d). Consequently, a 2152-bp deletion from IVS27-697 to IVS30 + 532 was confirmed (Fig. 3d). This deletion was also confirmed by MLPA (Fig. 3e).

Large deletion identified by LR-PCR. a LR-PCR products amplified from exons 22 to 34 in PKD1. The upper band is 10,524 bp as a normal allele, and the lower band is a shorter band by approximately 2 kb as a mutant allele. b The mean coverage in exons 27–30 of the patient with deletion mutation (no. 31) is much lower than those of two control patients (no. 21 and 41). c Nested PCR product amplified with IF271 and IR301 primers using the lower band of LR-PCR products amplified from exons 22 to 34 in PKD1 in patient no. 31 as a template. d PKD1 gene structure and the 2152-bp deletion in patient no. 31. e MLPA in patient no. 31. The counts in exons 27, 29, and 30 decrease to approximately 0.5 in patient no. 31
Among seven patients with frameshift mutations, four had insertions or deletions of more than 5 bp. A nonsense mutation in PKD2 exon 1 was also identified (Supplementary Table 5).
In the 15 patients for whom hybridization-based NGS could not be performed, PKD1 exhibited three frameshift mutations, one nonsense mutation, one splicing mutation, one in-frame deletion, and two substitutions, whereas PKD2 had two frameshift mutations and one splicing mutation (Fig. 2, Table 1, Supplementary Table 6).
Novel substitution evaluation
In addition to checking the PKDB, analyses using PKD Target were also performed. In the substitutions identified using hybridization-based NGS, 10 were reported in PKDB, whereas 15 were novel substitutions. Among these, 12 were novel substitutions with a total score ≥ 14, and were included as pathogenic mutations. Although three were novel substitutions with total scores < 14, their UCSC Vertebrate Conservation Scores indicated “1-Likely pathogenic”. Therefore, these were also included as a pathogenic mutation (Supplementary Table 4). In two substitutions identified using LR-PCR-based NGS, one was reported in PKDB, and the other was a novel substitution with a total score of more than 14, included as pathogenic mutations (Supplementary Table 6). Regarding the minor allele frequencies, no substitution was identified in 1000 genomes, but only one substitution (PKD1, c.12444G>C, p.Glu4148Asp) was identified in HGVD. However, from low frequency of this substitution (0.000415) and silico analysis described above, we judged this substitution as a pathogenic mutation.
Mutations in PKD1 exon 1 analysed by Sanger sequencing
PKD1 exons 1 and 42 have a known weak point in that their NGS coverage is low. Sanger sequencing of these exons was performed on the 15 patients whose mutations could not be identified using NGS. A frameshift mutation in exon 1 of PKD1 was identified in one patient (Fig. 2, Table 1, Supplementary Table 7).
Large rearrangements identified by MLPA
MLPA was performed on the 14 patients for whom NGS and PKD1 exon 1 analysis could not detect any mutations. Two patients had deletions in PKD1 exons 3 and 5, two had a large deletion in PKD1, and one had a large deletion in PKD2 (Fig. 2, Supplementary Fig. 2, Table 1, Supplementary Table 8). The deletion (patient no. 57) of 30 bp in exon 5 was confirmed using Sanger sequencing. For the deletion of PKD1 exons 11–46 found in patient no. 124 (Fig. 4a, b), LR-PCR of exons 2–46 detected an approximately 9-kb PCR product that was not found in the control (Fig. 4c). Using this product as a template, nested PCR was performed by setting primers 11F3 and 3′UTR-R2 at IVS10 and 3′UTR, respectively. The PCR product (approximately 200 bp) was sequenced (Fig. 4d). A 28,490-bp deletion from IVS10 to exon 46 (reference sequence no. 54453) was confirmed (Fig. 4b). The range of the total exon deletion in PKD2 found in patient no. 4 could not be identified.

Large deletion identified using MLPA in patient no. 124. a MLPA in patient no. 124. The counts from exons 11 to 46 decrease to approximately 0.5. bPKD1 structure and the 28,490-bp deletion in patient no. 124. c LR-PCR products amplified from exons 2 to 46 in PKD1. The approximately 9-kb band appeared as a mutant allele. d Sanger sequence of PCR product amplified with 11F3 and 3′UTR-R2 primers using the band of LR-PCR products amplified from exons 2 to 46 in PKD1 in patient no. 124 as a template. The left half sequence shows IVS10 sequence and the right half sequence shows 3′UTR sequence
In total, genetic mutations were identified in 102 of 111 patients, resulting in a final mutation detection rate of 91.9% (Fig. 2, Table 2).
Discussion
In this study, genetic analyses using NGS and MLPA were performed on 111 Japanese patients with ADPKD. Several studies have used NGS on the ADPKD causative genes PKD1 and PKD2. In the first study, a LR-PCR-based NGS had a detection rate of 63% [8]. Recent studies using similar methods have reported significantly increased detection rates [10]. A hybridization-based NGS has a detection rate of 70% [9]. Thus, we performed NGS analyses using both a hybridization-based and a LR-PCR-based target enrichment method. Consequently, a hybridization-based NGS had a mutation detection rate of 79.2%, which was lower than a LR-PCR-based NGS on Japanese patients that had a detection rate of 89.1% as previously reported [10]. In addition, the LR-PCR-based NGS identified new mutations in nine patients. Among those nine patients, seven had frameshifts. Thus, insertions and deletions leading to frameshifts may be easier to detect with a LR-PCR-based NGS than with a hybridization-based NGS. Although a hybridization-based NGS could not detect insertions and deletions of more than 3 bp, with a LR-PCR-based NGS, four out of seven were deletions of more than 5 bp. Improved efficiency of NGS equipment is thought to be major reason for the effectiveness of these analyses, but the detection rate using a hybridization-based NGS on insertions and deletions over 3 bp may be low. Using both methods, areas with an extremely low coverage were identified, such as PKD1 exons 1 and 42 as well as PKD2 exon 1, which may be the reason why these regions are GC-rich [26]. However, a PKD2 exon 1 mutation was identified in one patient using a LR-PCR-based NGS. This may be due to the improved efficiency of NGS equipment. According to the previous report [10] and this study, the 90% detection rate may be the limit of NGS. For the remaining approximately 10% patients, we need other strategies for identifying mutations, such as MLPA and Sanger sequencing for PKD1 exon 1.
As MLPA is particularly useful for detecting large rearrangements, it is frequently used in the genetic analysis of ADPKD [11, 12]. In this study, MLPA was performed using a PKD1 and PKD2 kit, and deletions from six patients were identified [6/111 patients (5.4%)] including the patient (No. 31) whose mutation was identified using LR-PCR. Five deletions identified using MLPA could not be identified using NGS alone. Thus, we suggest that MLPA is necessary in addition to NGS for mutation detection in ADPKD.
In conclusion, NGS has rapidly advanced in the last several years, and its momentum has pushed the detection rate of ADPKD to exceed 85%. Although MLPA has a detection rate of approximately 5%, it has the ability to raise the mutation detection rate of ADPKD and proved to be a useful tool in our study. In this way, the cost of using NGS can be comparatively lowered, and could be used by businesses to conduct genetic analyses. However, ADPKD patients would greatly benefit from genetic analyses capable of a detection rate of 100%. Therefore, development of novel genetic diagnostic techniques must be attempted while existing methods are improved to increase the rate of diagnosis by genetic analyses.
References
Mochizuki T, Tsuchiya K, Nitta K. Autosomal dominant polycystic kidney disease: recent advances in pathogenesis and potential therapies. Clin Exp Nephrol. 2013;17:317–26.
Cornec-Le Gall E, Audrezet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau E, Jousset P, Guillodo MP, Grall-Jezequel A, Saliou P, Ferec C, Le Meur Y. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol JASN. 2013;24:1006–13.
Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS, Investigators TT. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367:2407–18.
Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Perrone RD, Dandurand A, Ouyang J, Czerwiec FS, Blais JD, Investigators TT. Multicenter, open-label, extension trial to evaluate the long-term efficacy and safety of early versus delayed treatment with tolvaptan in autosomal dominant polycystic kidney disease: the TEMPO 4:4 Trial. Nephrol Dialysis Transplant. 2017. https://doi.org/10.1093/ndt/gfx043.
Phakdeekitcharoen B, Watnick TJ, Germino GG. Mutation analysis of the entire replicated portion of PKD1 using genomic DNA samples. J Am Soc Nephrol JASN. 2001;12:955–63.
Watnick TJ, Piontek KB, Cordal TM, Weber H, Gandolph MA, Qian F, Lens XM, Neumann HP, Germino GG. An unusual pattern of mutation in the duplicated portion of PKD1 is revealed by use of a novel strategy for mutation detection. Hum Mol Genet. 1997;6:1473–81.
Watnick T, Phakdeekitcharoen B, Johnson A, Gandolph M, Wang M, Briefel G, Klinger KW, Kimberling W, Gabow P, Germino GG. Mutation detection of PKD1 identifies a novel mutation common to three families with aneurysms and/or very-early-onset disease. Am J Hum Genet. 1999;65:1561–71.
Rossetti S, Hopp K, Sikkink RA, Sundsbak JL, Lee YK, Kubly V, Eckloff BW, Ward CJ, Winearls CG, Torres VE, Harris PC. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol JASN. 2012;23:915–33.
Qi XP, Du ZF, Ma JM, Chen XL, Zhang Q, Fei J, Wei XM, Chen D, Ke HP, Liu XZ, Li F, Chen ZG, Su Z, Jin HY, Liu WT, Zhao Y, Jiang HL, Lan ZZ, Li PF, Fang MY, Dong W, Zhang XN. Genetic diagnosis of autosomal dominant polycystic kidney disease by targeted capture and next-generation sequencing: utility and limitations. Gene. 2013;516:93–100.
Kinoshita M, Higashihara E, Kawano H, Higashiyama R, Koga D, Fukui T, Gondo N, Oka T, Kawahara K, Rigo K, Hague T, Katsuragi K, Sudo K, Takeshi M, Horie S, Nutahara K. Technical evaluation: identification of pathogenic mutations in PKD1 and PKD2 in patients with autosomal dominant polycystic kidney disease by next-generation sequencing and use of a comprehensive new classification system. PLoS ONE. 2016;11:e0166288.
Kozlowski P, Bissler J, Pei Y, Kwiatkowski DJ. Analysis of PKD1 for genomic deletion by multiplex ligation-dependent probe assay: absence of hot spots. Genomics. 2008;91:203–8.
Yu G, Qian X, Wu Y, Li X, Chen J, Xu J, Qi J. Analysis of gene mutations in PKD1/PKD2 by multiplex ligation-dependent probe amplification: some new findings. Ren Fail. 2015;37:366–71.
Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, Parfrey P, Cramer B, Coto E, Torra R, San Millan JL, Gibson R, Breuning M, Peters D, Ravine D. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol JASN. 2009;20:205–12.
Irazabal MV, Rangel LJ, Bergstralh EJ, Osborn SL, Harmon AJ, Sundsbak JL, Bae KT, Chapman AB, Grantham JJ, Mrug M, Hogan MC, El-Zoghby ZM, Harris PC, Erickson BJ, King BF, Torres VE. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol JASN. 2015;26:160–72.
Tsuchiya K, Komeda M, Takahashi M, Yamashita N, Cigira M, Suzuki T, Suzuki K, Nihei H, Mochizuki T. Mutational analysis within the 3′ region of the PKD1 gene in Japanese families. Mutat Res. 2001;458:77–84.
Hayashi T, Mochizuki T, Reynolds DM, Wu G, Cai Y, Somlo S. Characterization of the exon structure of the polycystic kidney disease 2 gene (PKD2). Genomics. 1997;44:131–6.
Audrezet MP, Cornec-Le Gall E, Chen JM, Redon S, Quere I, Creff J, Benech C, Maestri S, Le Meur Y, Ferec C. Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat. 2012;33:1239–50.
Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–4.
Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB, Group IUGVW. In silico analysis of missense substitutions using sequence-alignment based methods. Hum Mutat. 2008;29:1327–36.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–46.
Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–74.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6.
Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65.
Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H, Doi K, Shimizu M, Nakabayashi K, Aoki Y, Tsurusaki Y, Morishita S, Kawaguchi T, Migita O, Nakayama K, Nakashima M, Mitsui J, Narahara M, Hayashi K, Funayama R, Yamaguchi D, Ishiura H, Ko WY, Hata K, Nagashima T, Yamada R, Matsubara Y, Umezawa A, Tsuji S, Matsumoto N, Matsuda F. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61:547–53.
Kurashige M, Hanaoka K, Imamura M, Udagawa T, Kawaguchi Y, Hasegawa T, Hosoya T, Yokoo T, Maeda S. A comprehensive search for mutations in the PKD1 and PKD2 in Japanese subjects with autosomal dominant polycystic kidney disease. Clin Genet. 2015;87:266–72.
Acknowledgements
We express our sincere appreciation to all the patients, collaborating physicians, and other medical staff for their important contributions to the study. This study was supported in part by JSPS KAKENHI Grant Number JP 15K09279 and by a Grant-in-Aid for Intractable Renal Diseases Research, Research on rare and intractable diseases, Health and Labour Sciences Research Grants from the Ministry of Health, Labour and Welfare of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Toshio Mochizuki and Ken Tsuchiya received Travel fees and honoraria for lectures from Otsuka Pharmaceutical Co. Toshio Mochizuki and Hiroshi Kataoka belong to an endowed department sponsored by Otsuka Pharmaceutical Co, Chugai Pharmaceutical Co, Kyowa Hakko Kirin Co, MSD Co, and JMS Co.
Ethical approval
All procedures performed in this study were approved by the research ethics committee of Tokyo Women’s Medical University (No. 196B) in accordance with the 1964 Helsinki Declaration and its later amendments, or with comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Mochizuki, T., Teraoka, A., Akagawa, H. et al. Mutation analyses by next-generation sequencing and multiplex ligation-dependent probe amplification in Japanese autosomal dominant polycystic kidney disease patients. Clin Exp Nephrol 23, 1022–1030 (2019). https://doi.org/10.1007/s10157-019-01736-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-019-01736-3