
Abstract
Emerging evidence suggests that long non-coding RNAs (lncRNAs) are critical regulators of diverse biological processes, including adipogenesis. Despite being considered an ideal animal model for studying adipogenesis, little is known about the roles of lncRNAs in the regulation of rabbit preadipocyte differentiation. In the present study, visceral preadipocytes isolated from newborn rabbits were cultured in vitro and induced for differentiation, and global lncRNA expression profiles of adipocytes collected at days 0, 3, and 9 of differentiation were analyzed by RNA-seq. A total of 2066 lncRNAs were identified from nine RNA-seq libraries. Compared to protein-coding transcripts, lncRNA transcripts exhibited characteristics of a longer length and lower expression level. Furthermore, 486 and 357 differentially expressed (DE) lncRNAs were identified when comparing day 3 vs. day 0 and day 9 vs. day 3, respectively. Target genes of DE lncRNAs were predicted by the cis-regulating approach. Prediction of functions revealed that DE lncRNAs when comparing day 3 vs. day 0 were involved in gene ontology (GO) terms of developmental growth, growth, developmental cell growth, and stem cell proliferation, and involved in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of PI3K-Akt signaling pathway, fatty acid biosynthesis, and the insulin signaling pathway. The DE lncRNAs when comparing day 9 vs. day 3 were involved in GO terms that associated with epigenetic modification and were involved in the KEGG pathway of cAMP signaling pathway. This study provides further insight into the regulatory function of lncRNAs in rabbit visceral adipose and facilitates a better understanding of different stages of preadipocyte differentiation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Studies on the regulatory mechanisms of visceral adipocyte differentiation have long become the core of fat development research due to the excessive accumulation of visceral adipose that was reported to be associated with cardiovascular disease and hypertension (Fox et al. 2007; Kim et al. 2019; Pischon et al. 2008). As an economically important domestic animal, rabbits (Oryctolagus_cuniculus) have less fat deposit compared with other mammals, such as swine, cattle, and sheep. Due to the naturally low-fat deposition during rabbit growth, rabbits are an ideal model for studying visceral adipose development (Desando et al. 2013; Gong et al. 2014; Maneschi et al. 2013; Wang et al. 2015) and have important clinical value.
Adipose tissue is a complex, highly active metabolic, and endocrine organ (Kershaw and Flier 2004). At the cellular level, the development of adipose tissue is primarily due to an increase in number and size of adipose cells. Moreover, the process from preadipocyte to mature adipocyte was known as preadipocyte differentiation or adipogenesis, which is a key process for lipid accumulation (Kai et al. 2011). Although the exact genetic mechanisms of adipogenesis remain unclear, it is generally accepted that preadipocyte differentiation is a highly regulated process that involves several transcription factors, including peroxisome proliferator-activated receptor gamma (PPARG) (Tontonoz et al. 1994), CCAAT/enhancer-binding protein α (CEBPA) (Lin and Lane 1994), and fatty acid binding protein 4 (FABP4) (Deng et al. 2019).
Long non-coding RNAs (lncRNAs) are a group of endogenous RNA molecules with a total length of more than 200 nucleotides, and lack a functional open reading frame (ORF) (Batista and Chang 2013; Quinn and Chang 2016; Rinn and Chang 2012). In previous studies, large-scale sequencing has revealed that lncRNAs were involved in many biological processes, such as skeletal muscle development in rabbit (Kuang et al. 2018), intramuscular adipose generation in pigs (Miao et al. 2018), and bovine mammary gland development (Tong et al. 2017). In vitro, several lncRNAs including SRA (Xu et al. 2010), lnc-RAP-n (Sun et al. 2013a), ADINR (Xiao et al. 2015), and lnc-leptin (Lo et al. 2018) have been identified as critical regulators during preadipocyte differentiation in human and mouse.
In our previous study, we identified many lncRNAs in rabbit adipose tissue (Wang et al. 2018a). However, at the cellular level, the profile and function of lncRNAs in rabbit adipogenesis remain largely unknown. To investigate the roles of lncRNAs at the cellular level, visceral preadipocytes were isolated from rabbits and lncRNA expression profiles were explored at days 0, 3, and 9 during preadipocyte differentiation. In this study, we focused on the identification and characterization of lncRNAs in rabbit visceral preadipocyte and we detected dynamic changes in lncRNA expression during visceral preadipocyte differentiation. The results of this study provide further insight into the regulatory function of lncRNAs in rabbit visceral adipose and facilitate a better understanding of different stages of preadipocyte differentiation.
Materials and methods
Ethics statement
This study was performed in accordance with the guidelines of Good Experimental Practices adopted by the Institute of Animal Science (Sichuan Agricultural University, Chengdu, China). All surgical procedures involving rabbits were performed according to approved protocols of the Biological Studies Animal Care and Use Committee, Sichuan Province, China.
Isolation and induction of rabbit preadipocytes
Visceral preadipocytes were isolated from perirenal adipose tissue of newborn New Zealand rabbits under sterile conditions. Briefly, adipose tissues were digested with 0.01% collagenase I (Gibco, Carlsbad, CA, USA). Then, preadipocytes were seeded into 6-well plates at a density of 6 × 105 cells per plate in complete medium (DME/F12, supplemented with 10% fetal bovine serum [FBS]) (DME/F12 was obtained Gibco, Carlsbad, CA, USA; fetal bovine serum was from Zeta life, Menlo Park, CA, USA), and preadipocytes were cultured in a humidified incubator at 37 °C and 5% CO2. Upon reaching confluency (set to day 0), cells were induced by the addition of differentiation medium I (DME/F12 with 1 μM dexamethasone, 500 μM 1-methy1-3-iosbutylxanthine, 1.7 μM insulin, 10% FBS) (dexamethasone, 1-methy1-3-iosbutylxanthine, and insulin were from Solarbio, Beijing, China) for 72 h (day 3). Next, cells were cultured with differentiation medium II (DME/F12, 1.7 μM insulin, 10% FBS) for an additional 72 h and further cultured in complete medium until day 9 (day 9). To identify lipid accumulation in cells and obtain mature adipocytes, the accumulation of lipid droplets was measured by Oil Red O staining. Based on three different phenotypes of lipid accumulation (almost no lipid droplets, a small amount of ring-shaped lipid droplets, and a cluster of bigger lipid droplets), a total of nine cell samples were collected, and each interval time point contained three biological replicates. RT-qPCR was performed to determine the mRNA expression levels of adipogenic marker genes, including PPARG, CEBPA, and FABP4 (primer sequences are shown in Table S1), and the 2-ΔΔCt method was used to analyze the relative expression. The housekeeping gene GAPDH served as control.
Library preparation and sequencing
Total RNA was isolated using TRIzol reagent (Invitrogen, Hong Kong, China). The RNA purity, integrity, and concentration were determined by Nanodrop (ThermoFisher, Carlsbad, CA, USA), Agilent Bioanalyzer2100 system (Agilent Technologies, Carlsbad, CA, USA), and a Qubit 2.0 fluorometer (Life Technologies, Carlsbad, CA, USA), respectively. RNA samples with an optical density 260:280 ratio > 1.9 and integrity number value > 8.0 were selected for library construction. The library construction and sequencing of samples were performed by Haplox Genome Center (Jiangxi, China). Briefly, 1 μg RNA was used per sample and rRNA was removed using NR603-VAHTS Total RNA-Seq Library Prep Kit (Vazyme Biotech Co., Ltd Nanjing, China). First-strand cDNA was synthesized using First Strand Enzyme Mix (Vazyme Biotech Co., Ltd Nanjing, China). The second-strand of cDNA was synthesized using Maturity Strand Marking buffer (Vazyme Biotech Co., Ltd Nanjing, China). Subsequently, the USER enzyme was used to degrade the cDNA strands that contained U instead of T. All purified libraries were sequenced on an Illumina Hiseq X platform, and then 150-bp paired-end reads were generated.
Transcript assembly and lncRNA identification
Raw reads in FASTQ format were analyzed and filtered using Fastp software (Chen et al. 2018) with default parameters. After removing the adapter sequences and low-quality reads, clean reads were obtained and mapped to the rabbit reference genome OryCun2.0 (ftp://ensembl.org/pub/release95/fasta/oryctolagus_cuniculus/) together with the genome annotation (OryCun2.0.95.gtf) using an aligner Histat2 (Kim et al. 2015), with parameters of “--rna-strandness RF” and “-dta”. Transcripts of each sample were initially assembled using the Stringtie program (Pertea et al. 2016). Then, transcripts from nine samples were merged to create a consensus transcriptome. The fragment per kilobase of transcript per million mapped reads (FPKM) and raw counts that mapped to corresponding transcripts were estimated using the Stringtie program (Pertea et al. 2016) with the parameter of “-e -B” (Pertea et al. 2016). To identify lncRNA candidates and classify lncRNAs based on location, merged transcripts were compared to the rabbit reference transcripts using Gffcampare software (Trapnell et al. 2012) and only transcripts that were marked with either an “x” (exonic overlap with a reference on the opposite strand, potential anti-sense lncRNA), “u” (unknown intergenic transcript, potential intergenic lncRNA), “o” (exonic overlap with a reference on the sense strand, potential sense lncRNA), or “i” (a transfrag falling entirely within a reference intron, potential intronic lncRNA) were retained (Kuang et al. 2018; Wu et al. 2016). The identification pipeline of lncRNAs consisted of two steps: transcript basic filtration and potential coding ability filtration. First, transcripts were selected that had a length of > 200 bp, contained ≥ 2 exons, and displayed a FPKM of > 0.5 through basic filtration. Subsequently, CPC (Kong et al. 2007), CNCI (Sun et al. 2013b), CPAT (Wang et al. 2013), and Pfam (Finn et al. 2014) were used to identify putative lncRNAs by analyzing their coding potential, and only those simultaneously identified by all these four different types of software were considered credible lncRNAs.
Differentially expressed lncRNAs and functional enrichment analysis
To identify lncRNAs that are associated with the differentiation of rabbit preadipocytes, lncRNA expression profiles of RNA-Seq libraries were compared according to the different stages of preadipocyte differentiation. The raw counts that yielded from the Stringtie program (Pertea et al. 2016) were applied in differential expression analysis using R package Deseq2 (Love et al. 2014). LncRNA transcripts with a threshold of Padj < 0.05 and |log2Fold-Change)| > 1 were considered as differentially expressed (DE) lncRNAs. All DE lncRNAs in nine libraries were clustered using the Python module Seaborn (Hunter 2007). Coding genes located at DE lncRNA within 100 kb were considered potential cis-regulated target genes. The prediction of DE lncRNAs functions was based on the functional annotation of the potential cis-regulated target gene. Gene ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for the candidate target gene were performed using the David database (Da et al. 2009).
RT-qPCR for validation of DE lncRNAs
The RT-qPCR primers for the validation of DE lncRNAs were designed by online software Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) (Table S1). Total RNA was converted to cDNA using the PrimeScripts RT Reagent Kit containing gDNA Eraser (TAKARA, Dalian, China). The qPCR was determined using a SYBR II master mix kit (TAKARA, Dalian, China) and performed on a Bio-Rad CFX manager according to the manufacturer’s instructions. The amplification reaction program was as follows: 95 °C for 10 s, followed by 40 cycles of 95 °C for 5 s, and 20 s at the melting temperature (Tm). Melting curve analysis was performed from 55 to 95 °C with increments of 0.5 °C, and the 2-ΔΔCt method was used to analyze relative expression. The housekeeping gene GAPDH served as control.
Statistical analyses
Statistical analysis was performed using SPSS Statistics 20.0 (SPSS Inc., Chicago, IL, USA).
Results
Differentiation of visceral preadipocytes
Adipocytes were stained with Oil Red O on day 0, day 3, and day 9 after induction of differentiation (Fig. 1a). As shown in Fig. 1b, different amounts of lipids accumulated within these three staged adipocytes, showing almost no lipid droplets on day 0, a small amount of ring-shaped lipid droplets on day 3, and a cluster of bigger lipid droplets on day 9. The expression level of adipogenic marker genes showed a significant increase over the course of induced differentiation of the preadipocytes, reached the highest level on day 5 (7-fold increase for PPARG, 23-fold increase for CEBPA, and 59-fold increase for FABP4), and then decreased on day 7 and day 9 (Fig. 1c, 1d, 1e).

Differentiation of rabbit visceral preadipocytes. a Oil red O staining of lipid droplets in cells that were induced differentiation at day 0, day 3, and day 9. b Identification of mature adipocyte with Oil red O staining, 50 μm. c–e The relative expression level of adipogenic maker genes PPARG, CEBPA, and FABP4. GAPDH served as a control. The data shows the means of three independent experiments, *P < 0.05, **P < 0.01, ***P < 0.001 versus day 0
Overview of RNA sequencing
A total of nine RNA-seq libraries were constructed from rabbit preadipocyte cells that underwent induced differentiation of 0 day, 3 days, and 9 days, and three biological repetitions were set for per stage. An average of 18.94 Gb pair-end raw reads in all nine samples were generated from these libraries. After quality control, a total of 1,109,630,370 clean reads (155.03 Gb) were obtained. The Q20 (percentage of reads with a Phred quality value > 20) and Q30 (percentage of reads with a Phred quality value > 30) of clean reads ranged from 97.01 to 97.50% and 92.40 to 93.53%, respectively. The mapped rate of nine samples ranged from 90.67 to 91.77% (Table S2).
Identification and characterization of lncRNAs
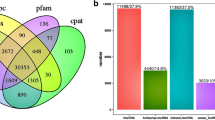
The lncRNA-set contained 2878, 3032, 2864, and 2809 putative lncRNA transcripts and was predicted by CPC2, CNCI, CPAT, and Pfam, respectively. The intersection of these lncRNA-sets yielded 2066 credible lncRNAs (Fig. 2a). These lncRNAs included 103 sense lncRNAs, 878 intronic lncRNAs, 976 intergenic lncRNAs, and 109 anti-sense lncRNAs (Fig. 2b). The ORF length size of most lncRNA transcripts was less than 100 (Fig. 2c). When compared to protein-coding RNAs, lncRNAs were found to be longer in transcript length (Fig. 2d) and lower in expression level (Fig. 2e).

Identification of lncRNAs during the rabbit visceral preadipocyte differentiation and comparing of lncRNA transcripts to protein-coding transcripts. a The Venn diagram of lncRNA transcripts which yield from four types software of CPC2, CPAT, CNCI, and Pfam. Only the lncRNA identified by all the four types of software were used for downstream analyses. b Classification of identified lncRNAs. c The ORF length distribution of lncRNA transcripts. d Transcripts length distribution of lncRNA transcripts and mRNA transcripts. e Expression analysis of lncRNAs and protein-coding RNAs
Screening and clustering of differentially expressed lncRNAs
Dynamic changes of lncRNA expression during rabbit preadipocyte differentiation were detected using Deseq2. When comparing day 3 vs. day 0 (Fig. 3a and Table S3) and day 9 vs. day 3 (Fig. 3b and Table S4), 486 DE lncRNAs (219 up-regulated, 267 down-regulated) and 357 DE lncRNAs (208 up-regulated, 149 down-regulated) were detected, respectively. Venn diagram analysis showed 242 DE lncRNAs that identified in the day 3 vs. day 0 also differentially expressed in the day 9 vs. day 3 (Fig. 3c). The expression level (FPKM) of the union set of 601 DE lncRNAs in the two comparisons (day 3 vs. day 0 and day 9 vs. day 3) showed a similar pattern across different libraries at the same stage, and the expression pattern of DE lncRNAs between day 0 and day 9 was similar (Fig. 3d and Table S5).

Analysis of DE lncRNA during rabbit preadipocyte differentiation. a, b Vocalo plot analysis of DE lncRNAs in the comparison day 3 vs. day 0 and day 9 vs. day 3, red refer to up-regulated lncRNA and blue refer to down-regulated lncRNA (|log2(Fold_Change)| > 1 and Padj < 0.05). c Venn diagram of showing the number of DE lncRNAs. d Hierarchical clustering analysis of DE lncRNA expression profiles based on FPKM, red refer to higher expression level, and blue refer to lower expression level
Functional enrichment analysis when comparing day 3 vs. day 0 of the differentiation
Between day 0 and day 3 of rabbit visceral preadipocyte differentiation, the prediction of cis-regulated target genes showed that 399 out of 486 DE lncRNAs corresponded to 656 protein-coding RNAs. GO enrichment results showed that 118 GO terms were significantly enriched, and the top 10 significantly enriched GO terms in the categories biological processes (BP), cellular components (CC), and molecular functions (MF) are presented in Fig. 4. In addition, several GO terms associated with preadipocyte proliferation or differentiation such as stem cell proliferation, growth, positive regulation of cellular biosynthetic process, and developmental cell growth were significantly enriched (Table S6). KEGG pathway analysis showed that 25 pathways were significantly enriched, and the top 20 significantly enriched pathways are presented in Fig. 5. Several pathways associated with preadipocyte differentiation, such as the PI3K-Akt signaling pathway, fatty acid biosynthesis, and insulin signaling pathway were also significantly enriched (Table S7).

GO enrichment of the cis-regulated target genes of DE lncRNAs when comparing day 3 vs. day 0 during rabbit adipogenesis, with showing the top 10 significantly enriched GO terms in BP, CC, and MF (P < 0.05)

KEGG pathway analysis of the cis-regulated target genes of DE lncRNAs when comparing day 3 vs. day 0 during rabbit adipogenesis, with showing the top 20 significantly enriched pathways (P < 0.05)
Functional enrichment analysis when comparing day 9 vs. day 3 of the differentiation
Between day 3 and day 9 of rabbit visceral preadipocyte differentiation, the prediction of cis-regulated target genes showed that 293 out of 357 DE lncRNAs corresponded to 558 protein-coding RNAs. GO enrichment results showed that 119 GO terms were significantly enriched, and the top 10 significantly enriched GO terms in the categories BP, CC, and MF are presented in Fig. 6. Furthermore, several GO terms associated with preadipocyte differentiation, such as regulation of cell differentiation, developmental cell growth, and positive regulation of protein phosphorylation, were significantly enriched (Table S8). In addition, the GO terms associated with epigenetic modification, such as chromosomal part, chromosomal region, and H4/H2A histone acetyltransferase complex, were also significantly enriched. KEGG pathway analysis showed that 12 pathways were significantly enriched, including the cAMP signaling pathway (Table S9), and all the 12 significantly enriched pathways are presented in Fig. 7.

GO enrichment of the cis-regulated target genes of DE lncRNAs when comparing day 9 vs. day 3 during rabbit adipogenesis, with showing the top 10 significantly enriched GO terms in BP, CC, and MF (P < 0.05)

KEGG pathway analysis of the cis-regulated target genes of DE lncRNAs when comparing day 9 vs. day 3 during rabbit adipogenesis, with showing the significantly enriched pathways (P < 0.05)
Validation of DE lncRNAs
To validate the RNA-seq results, we randomly selected six DE lncRNAs and evaluated their expression level at the initial stage or maturity stage of adipogenesis by RT-qPCR. The RT-qPCR results demonstrated that all six lncRNAs (MSTRG.1657.2, MSTRG.2112.1, MSTRG.6545.1, MSTRG.7207.5, MSTRG.13853.1, and MSTRG.16280.4) were differentially expressed when comparing day 3 vs. day 0 or day 9 vs. day 3. In addition, the expression patterns were similar to the RNA-seq results (Fig 8). Therefore, lncRNA expression levels as demonstrated in this study by RNA-seq are reliable.

Validation of six randomly selected DE lncRNAs by RT-qPCR
Discussion
In this study, rabbit preadipocytes were induced for differentiation using the well-known induction cocktail DEX-IBMX-insulin (DMI) (Zebisch et al. 2012). The expression level of adipogenesis-related transcriptional factor genes PPARG, CEBPA, and FABP4 significantly increased after preadipocytes were induced for differentiation and many of big lipid droplets were observed after mature adipocytes were stained by Oil red O on day 9. Together, these results indicated that the rabbit adipocytes model was successfully established.
LncRNAs were explored according to the structure and protein-coding ability for the assembled transcripts using bioinformatics analysis. To obtain credible lncRNAs, we calculated the intersection of non-coding results that yielded from four types of software of CPC2, CNCI, CPAT, and Pfam. In this study, a total of 2066 lncRNAs identified in rabbit visceral preadipocytes during differentiation. When compared to protein-coding RNA, the lncRNAs identified in visceral preadipocytes showed common characteristic of lower expression levels with those identified in other species, such as pigs (Huang et al. 2018), goats (Bakhtiarizadeh and Salami 2019), and chickens (Zhang et al. 2017), but were relatively longer in transcript length. The longer length of lncRNAs when compared to protein-coding RNAs might be attributed to the poor conservation of lncRNA structure among different species (Johnsson et al. 2014). Rabbit lncRNAs with longer length might tend to acquire complex secondary and tertiary structures, whereas links between these complex structure and function are still not well defined. To our knowledge, lncRNA structure might be associated with the rapid rate of evolution (Ulitsky and Bartel 2013), but whether the longer length of lncRNA is related to evolution in rabbit requires more systematic sequence analysis among different species.
In this study, a considerable number of lncRNAs participated in the differentiation of rabbit preadipocytes. A total of 486 and 357 DE lncRNAs were detected when comparing of day 3 vs. day 0 and day 9 vs. day 3, respectively. The number of DE lncRNAs at the initial stage was higher compared to the maturity stage. The result was similar to a study of lncRNA expression during the differentiation of preadipocytes in chickens (Zhang et al. 2017). At the initial stage of preadipocytes induced differentiation, cells changed from proliferative state to differentiation state, and this process was accompanied by fading of stem cell pluripotency and the generation of lipids (Ding et al. 2015), and thereby might require a more complex regulation of multiple lncRNAs. However, at the maturity stage of induced differentiation, most cells were in the state of differentiation and the principal change involved lipid accumulation; this monotony of changes in cell state might require the regulation of relatively less lncRNAs. In this study, the expression pattern of lncRNA in mature adipocytes was close to that of undifferentiated preadipocytes, whereas 486 and 357 DE lncRNAs were detected on the day 3 vs. day 0 and day 9 vs. day 3, thereby indicating that some lncRNAs might act on specific stages during rabbit adipogenesis. To our knowledge, non-coding RNAs could play regulatory role in specific stages of adipogenesis (Liu et al. 2011; Peng et al. 2013), including a lncRNA of slincRAD identified in early adipogenesis in mouse (Yi et al. 2019); our DE lncRNAs identified in rabbit suggested that some lncRNAs might specifically play roles in the initiation of differentiation. Taken together, the DE lncRNAs identified in our study might be important regulators of preadipocyte differentiation. Most lncRNAs present in current databases have not yet been annotated, and studies have suggested that lncRNA expression could act on neighboring protein-coding genes by a cis-regulating approach (Ren et al. 2016; Wang et al. 2016). To investigate DE lncRNA functions in rabbit visceral preadipocyte differentiation, we predicted potential cis-regulation target genes based on the distance of lncRNA to protein-coding genes. Subsequently, GO enrichment and KEGG pathway analysis were performed based on the cis-regulated targets.
Between the day 0 and day 3 of rabbit preadipocyte differentiation, cis-regulated target genes of the DE lncRNAs were principally enriched in GO terms that were associated with preadipocyte cell growth or differentiation, such as positive regulation of cellular metabolic process, developmental growth, growth, stem cell proliferation, and developmental cell growth. The enrichment of these GO terms indicated that a percentage of cells was already differentiated, and a percentage of cells were still proliferating in the initial stage of preadipocyte differentiation. Protein phosphorylation plays an important role in adipogenesis (Mota de Sa et al. 2017). Protein phosphorylation changes of PPARG, CEBPA, and STAT have been reported to regulate the expression of downstream adipogenesis genes and modulate adipogenesis (Yang et al. 2018). The enrichment of GO terms, such as positive regulation of protein phosphorylation, positive regulation of tyrosine phosphorylation of Stat5 protein, and positive regulation of tyrosine phosphorylation of Stat5 protein (Table S6), suggested that DE lncRNAs might regulate rabbit adipogenesis via modulating protein phosphorylation. KEGG pathway analysis showed that cis-target genes of the DE lncRNAs when comparing day 3 vs. day 0 were involved in the PI3K-Akt signaling pathway, fatty acid biosynthesis, and insulin signaling pathway (Table S7). The PI3K-Akt signaling pathways have been found as a critical pathway in adipogenesis (Song et al. 2017; Wang et al. 2018b, 2019), and in a recent study, it was shown that lncRNA lnc-ORA (Cai et al. 2019) was involved in PI3K-Akt signaling pathway in 3T3-L1 adipogenesis. Our DE lncRNAs enriched in PI3K-Akt signaling pathway suggested that these DE lncRNAs might be a regulator in the early differentiation of rabbit preadipocytes (28 DE lncRNAs and their target genes are listed in Table S10). As expected, the enrichment pathway of the fatty acid biosynthesis signal pathway explained the formation of lipids in our study and indicated that some DE lncRNAs might regulate genes involved in fatty acid biosynthesis via cis-regulation (six lncRNAs and their target genes are listed in Table S10). The insulin signaling pathway is also a well-known pathway in adipogenesis (Lee 2017). Our DE lncRNAs enriched in the insulin signaling pathway suggested that these DE lncRNAs might regulate rabbit adipogenesis via the insulin signaling pathway by cis-regulating (10 lncRNAs and their target genes are listed in Table S10).
Previous studies have reported that lncRNAs could modulate cell processes via chromatin remodeling or histone modification (Hacisuleyman et al. 2014; Huang et al. 2017; Wei et al. 2016). Between the day 3 and day 9 of rabbit preadipocyte differentiation, cis-regulated target genes of DE lncRNAs were principally enriched in the GO terms of cellular component category associated chromatin remodeling and histone modification, such as the chromosomal part, chromosome, nuclear chromosome part, nuclear chromosome, chromosomal region, H4/H2A histone acetyltransferase complex, NuA4 histone acetyltransferase complex, or H4 histone acetyltransferase complex. Enrichment of those GO terms indicated the possible roles of the DE lncRNAs in regulating rabbit adipogenesis via chromatin remodeling and histone modification at the maturity stage. Several GO terms including the positive regulation of cellular metabolic process, developmental cell growth, and positive regulation of protein phosphorylation were enriched. These GO terms were simultaneously enriched by the DE lncRNAs in day 3 vs. day 0 and those in day 9 vs. day 3, which may suggest similar ways in regulating adipogenesis in both the early and late stages of rabbit preadipocyte differentiation. The cAMP signaling pathway has been reported to play a key role in regulating adipogenesis in mouse (Billert et al. 2018; Lee et al. 2018; Rogne and Tasken 2014). Our DE lncRNAs enriched in the KEGG pathway of cAMP signaling pathway suggested that some DE lncRNAs might regulate rabbit adipogenesis via the cAMP signaling pathway between day 3 and day 9 (11 lncRNAs and their targets are listed in Table S10).
Conclusions
Based on the analysis of the lncRNAs transcriptome of three stages during differentiation in rabbit visceral preadipocytes, a total of 2066 lncRNAs were identified in rabbit visceral preadipocytes by RNA-seq, and a total of 486 and 357 lncRNAs were differentially expressed when comparing day 3 vs. day 0 and day 9 vs. day 3, respectively. Some DE lncRNAs were found to be involved in the critical KEGG pathways that have been extensively studied in adipogenesis in other species. Those lncRNAs might modulate the rabbit preadipocyte differentiation and lipid accumulation through cis-regulating. Taken together, in this study, we provided a valuable resource for further exploring of rabbit lncRNA and to facilitate a better understanding of preadipocyte differentiation in the rabbit.
References
Bakhtiarizadeh MR, Salami SA (2019) Identification and expression analysis of long noncoding RNAs in fat-tail of sheep breeds. G3 (Bethesda) 9:1263–1276
Batista PJ, Chang HY (2013) Long noncoding RNAs: cellular address codes in development and disease. Cell 152:1298–1307
Billert M, Wojciechowicz T, Jasaszwili M, Szczepankiewicz D, Wasko J, Kazmierczak S, Strowski MZ, Nowak KW, Skrzypski M (2018) Phoenixin-14 stimulates differentiation of 3T3-L1 preadipocytes via cAMP/Epac-dependent mechanism. Biochim Biophys Acta Mol Cell Biol Lipids 1863:1449–1457
Cai R, Tang G, Zhang Q, Yong W, Zhang W, Xiao J, Wei C, He C, Yang G, Pang W (2019) A novel lnc-RNA, named lnc-ORA, is identified by RNA-Seq analysis, and its knockdown inhibits adipogenesis by regulating the PI3K/AKT/mTOR signaling pathway. Cells 8(5):E477
Chen S, Zhou Y, Chen Y, Gu J (2018) fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884–i890
Da WH, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57
Deng T, Wang Y, Wang C, Yan H (2019) FABP4 silencing ameliorates hypoxia reoxygenation injury through the attenuation of endoplasmic reticulum stress-mediated apoptosis by activating PI3K/Akt pathway. Life Sci 224:149–156
Desando G, Cavallo C, Sartoni F, Martini L, Parrilli A, Veronesi F, Fini M, Giardino R, Facchini A, Grigolo B (2013) Intra-articular delivery of adipose derived stromal cells attenuates osteoarthritis progression in an experimental rabbit model. Arthritis Res Ther 15:R22
Ding F, Li QQ, Li L, Gan C, Yuan X, Gou H, He H, Han CC, Wang JW (2015) Isolation, culture and differentiation of duck (Anas platyrhynchos) preadipocytes. Cytotechnology 67:773–781
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J (2014) Pfam: the protein families database. Nucleic Acids Res 42:222–230
Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, Vasan RS, Murabito JM, Meigs JB, Cupples LA, D’Agostino RB Sr, O’Donnell CJ (2007) Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation 116:39–48
Gong L, Wang C, Li Y, Sun Q, Li G, Wang D (2014) Effects of human adipose-derived stem cells on the viability of rabbit random pattern flaps. Cytotherapy 16:496–507
Hacisuleyman E, Goff LA, Trapnell C, Williams A, Henao-Mejia J, Sun L, Mcclanahan P, Hendrickson DG, Sauvageau M, Kelley DR (2014) Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat Struct Mol Biol 21:198–206
Huang Y, Jin C, Zheng Y, Li X, Zhang S, Zhang Y, Jia L, Li W (2017) Knockdown of lncRNA MIR31HG inhibits adipocyte differentiation of human adipose-derived stem cells via histone modification of FABP4. Sci Rep 7:8080
Huang W, Zhang X, Li A, Xie L, Miao X (2018) Genome-wide analysis of mRNAs and lncRNAs of intramuscular fat related to lipid metabolism in two pig breeds. Cell Physiol Biochem 50:2406–2422
Hunter JD (2007) Matplotlib: a 2D graphics environment. Comput Sci Eng 9:90–95
Johnsson P, Lipovich L, Grander D, Morris KV (2014) Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta 1840:1063–1071
Kai S, Kusminski CM, Scherer PE (2011) Adipose tissue remodeling and obesity. J Clin Invest 121:2094
Kershaw EE, Flier JS (2004) Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89:2548–2556
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360
Kim HJ, Kwon H, Jeong SM, Hwang SE, Park JH (2019) Effects of abdominal visceral fat compared with those of subcutaneous fat on the association between PM10 and hypertension in Korean men: a cross-sectional study. Sci Rep 9:5951
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G (2007) CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35:W345–W349
Kuang L, Lei M, Li C, Zhang X, Ren Y, Zheng J, Guo Z, Zhang C, Yang C, Mei X, Fu M, Xie X (2018) Identification of long non-coding RNAs related to skeletal muscle development in two rabbit breeds with different growth rate. Int J Mol Sci 19:E2046
Lee MJ (2017) Hormonal regulation of adipogenesis. Compr Physiol 7:1151–1195
Lee HL, Qadir AS, Park HJ, Chung E, Lee YS, Woo KM, Ryoo HM, Kim HJ, Baek JH (2018) cAMP/protein kinase A signaling inhibits Dlx5 expression via activation of CREB and subsequent C/EBPbeta induction in 3T3-L1 preadipocytes. Int J Mol Sci 19:E3161
Lin FT, Lane MD (1994) CCAAT/enhancer binding protein alpha is sufficient to initiate the 3T3-L1 adipocyte differentiation program. Proc Natl Acad Sci U S A 91:8757–8761
Liu S, Yang Y, Wu J (2011) TNFalpha-induced up-regulation of miR-155 inhibits adipogenesis by down-regulating early adipogenic transcription factors. Biochem Biophys Res Commun 414:618–624
Lo KA, Huang S, Walet ACE, Zhang ZC, Leow MK, Liu M, Sun L (2018) Adipocyte long-noncoding RNA transcriptome analysis of obese mice identified Lnc-leptin, which regulates leptin. Diabetes 67:1045–1056
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Maneschi E, Vignozzi L, Morelli A, Mello T, Filippi S, Cellai I, Comeglio P, Sarchielli E, Calcagno A, Mazzanti B, Vettor R, Vannelli GB, Adorini L, Maggi M (2013) FXR activation normalizes insulin sensitivity in visceral preadipocytes of a rabbit model of MetS. J Endocrinol 218:215–231
Miao Z, Wang S, Zhang J, Wei P, Guo L, Liu D, Wang Y, Shi M (2018) Identification and comparison of long non-conding RNA in Jinhua and Landrace pigs. Biochem Biophys Res Commun 506:765–771
Mota de Sa P, Richard AJ, Hang H, Stephens JM (2017) Transcriptional regulation of adipogenesis. Compr Physiol 7:635–674
Peng Y, Xiang H, Chen C, Zheng R, Chai J, Peng J, Jiang S (2013) MiR-224 impairs adipocyte early differentiation and regulates fatty acid metabolism. Int J Biochem Cell Biol 45:1585–1593
Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL (2016) Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11:1650–1667
Pischon T, Boeing H, Hoffmann K, Bergmann M, Schulze MB, Overvad K, van der Schouw YT, Spencer E, Moons KG, Tjonneland A, Halkjaer J, Jensen MK, Stegger J, Clavel-Chapelon F, Boutron-Ruault MC, Chajes V, Linseisen J, Kaaks R, Trichopoulou A, Trichopoulos D, Bamia C, Sieri S, Palli D, Tumino R, Vineis P, Panico S, Peeters PH, May AM, Bueno-de-Mesquita HB, van Duijnhoven FJ, Hallmans G, Weinehall L, Manjer J, Hedblad B, Lund E, Agudo A, Arriola L, Barricarte A, Navarro C, Martinez C, Quiros JR, Key T, Bingham S, Khaw KT, Boffetta P, Jenab M, Ferrari P, Riboli E (2008) General and abdominal adiposity and risk of death in Europe. N Engl J Med 359:2105–2120
Quinn JJ, Chang HY (2016) Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17:47–62
Ren H, Wang G, Chen L, Jiang J, Liu L, Li N, Zhao J, Sun X, Zhou P (2016) Genome-wide analysis of long non-coding RNAs at early stage of skin pigmentation in goats (Capra hircus). BMC Genomics 17:67
Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81:145–166
Rogne M, Tasken K (2014) Compartmentalization of cAMP signaling in adipogenesis, lipogenesis, and lipolysis. Horm Metab Res 46:833–840
Song F, Jiang D, Wang T, Wang Y, Lou Y, Zhang Y, Ma H, Kang Y (2017) Mechanical stress regulates osteogenesis and adipogenesis of rat mesenchymal stem cells through PI3K/Akt/GSK-3beta/beta-catenin signaling pathway. Biomed Res Int 2017:6027402
Sun L, Goff LA, Trapnell C, Alexander R, Lo KA, Hacisuleyman E, Sauvageau M, Tazonvega B, Kelley DR, Hendrickson DG (2013a) Long noncoding RNAs regulate adipogenesis. Proc Natl Acad Sci U S A 110:3387–3392
Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C, Liu Y, Chen R, Zhao Y (2013b) Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res 41:e166
Tong C, Chen Q, Zhao L, Ma J, Ibeagha-Awemu EM, Zhao X (2017) Identification and characterization of long intergenic noncoding RNAs in bovine mammary glands. BMC Genomics 18:468
Tontonoz P, Hu E, Spiegelman BM (1994) Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 79:1147–1156
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578
Ulitsky I, Bartel DP (2013) lincRNAs: genomics, evolution, and mechanisms. Cell 154:26–46
Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W (2013) CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res 41:e74
Wang W, He N, Feng C, Liu V, Zhang L, Wang F, He J, Zhu T, Wang S, Qiao W, Li S, Zhou G, Zhang L, Dai C, Cao W (2015) Human adipose-derived mesenchymal progenitor cells engraft into rabbit articular cartilage. Int J Mol Sci 16:12076–12091
Wang Y, Xue S, Liu X, Liu H, Hu T, Qiu X, Zhang J, Lei M (2016) Analyses of long non-coding RNA and mRNA profiling using RNA sequencing during the pre-implantation phases in pig endometrium. Sci Rep 6:20238
Wang GZ, Du K, Hu SQ, Chen SY, Jia XB, Cai MC, Shi Y, Wang J, Lai SJ (2018a) Genome-wide identification and characterization of long non-coding RNAs during postnatal development of rabbit adipose tissue. Lipids Health Dis 17:271
Wang X, Chen J, Rong C, Pan F, Zhao X, Hu Y (2018b) GLP-1RA promotes brown adipogenesis of C3H10T1/2 mesenchymal stem cells via the PI3K-AKT-mTOR signaling pathway. Biochem Biophys Res Commun 506:976–982
Wang N, Li Y, Li Z, Ma J, Wu X, Pan R, Wang Y, Gao L, Bao X, Xue P (2019) IRS-1 targets TAZ to inhibit adipogenesis of rat bone marrow mesenchymal stem cells through PI3K-Akt and MEK-ERK pathways. Eur J Pharmacol 849:11–21
Wei S, Du M, Jiang Z, Hausman G, Zhang L, Dodson M (2016) Long noncoding RNAs in regulating adipogenesis: new RNAs shed lights on obesity. Cell Mol Life Sci 73:1–9
Wu Y, Cheng T, Liu C, Liu D, Zhang Q, Long R, Zhao P, Xia Q (2016) Systematic identification and characterization of long non-coding RNAs in the silkworm, Bombyx mori. PLoS One 11:e0147147
Xiao T, Liu L, Li H, Yu, Luo (2015) Long noncoding RNA ADINR regulates adipogenesis by transcriptionally activating C/EBPα. Stem Cell Rep 5:856–865
Xu B, Gerin I, Miao H, Dang VP, Johnson CN, Xu R, Chen XW, Cawthorn WP, Macdougald OA, Koenig RJ (2010) Multiple roles for the non-coding RNA SRA in regulation of adipogenesis and insulin sensitivity. PLoS One 5:e14199
Yang SM, Park YK, Kim JI, Lee YH, Lee TY, Jang BC (2018) LY3009120, a pan-Raf kinase inhibitor, inhibits adipogenesis of 3T3-L1 cells by controlling the expression and phosphorylation of C/EBP-alpha, PPAR-gamma, STAT3, FAS, ACC, perilipin A, and AMPK. Int J Mol Med 42:3477–3484
Yi F, Zhang P, Wang Y, Xu Y, Zhang Z, Ma W, Xu B, Xia Q, Du Q (2019) Long non-coding RNA slincRAD functions in methylation regulation during the early stage of mouse adipogenesis. RNA Biol 16:1401–1413
Zebisch K, Voigt V, Wabitsch M, Brandsch M (2012) Protocol for effective differentiation of 3T3-L1 cells to adipocytes. Anal Biochem 425:88–90
Zhang T, Zhang X, Han K, Zhang G, Wang J, Xie K, Xue Q (2017) Genome-wide analysis of lncRNA and mRNA expression during differentiation of abdominal preadipocytes in the chicken. G3 (Bethesda) 7:953–966
Funding
This work was supported by the earmarked fund for China Agriculture Research System (CARS-44-A-2).
Author information
Authors and Affiliations
Contributions
Conceptualization: Song-Jia Lai.
Formal analysis: Kun Du, Guo-Ze Wang.
Resource: An-yong Ren, Ming-cheng Cai, Gang Luo, Xian-bo Jia, Shen-qiang Hu, Jie Wang, Shi-Yi Chen, Song-Jia Lai.
Writing: Kun Du.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Table S1.
Primers of genes used in RT-qPCR (docx format). (DOCX 15 kb)
Table S2.
Summary of RNA-seq data and reads mapping (docx format). (DOCX 16 kb)
Table S3.
Differentially expressed analysis when comparing day 3 vs. day 0 (xlsx format). (XLSX 50 kb)
Table S4.
Differentially expressed analysis when comparing day 9 vs. day 3 (xlsx format). (XLSX 39 kb)
Table S5.
FPKM of lncRNA transcripts (xlsx format). (XLSX 209 kb)
Table S6.
Result of GO enrichment between day 0 and day 3 of rabbit adipogenesis (xlsx format). (XLSX 23 kb)
Table S7.
Result of KEGG pathway analysis between day 0 and day 3 of rabbit adipogenesis (xlsx format). (XLSX 12 kb)
Table S8.
Result of GO enrichment between day 3 and day 9 of rabbit adipogenesis (xlsx format). (XLSX 22 kb)
Table S9.
Result of KEGG pathway analysis between day 3 and day 9 of rabbit adipogenesis (xlsx format). (XLSX 11 kb)
Table S10.
DE LncRNAs in key pathways that might regulate rabbit adipogenesis (xlsx format). (XLSX 10 kb)
Rights and permissions
About this article

Cite this article
Du, K., Wang, GZ., Ren, Ay. et al. Genome-wide identification and characterization of long non-coding RNAs during differentiation of visceral preadipocytes in rabbit. Funct Integr Genomics 20, 409–419 (2020). https://doi.org/10.1007/s10142-019-00729-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-019-00729-5