
Abstract
Plant stress response is a complex molecular process based on transcriptional and posttranscriptional regulation of many stress-related genes. microRNAs are the best-studied class of small RNAs known to play key regulatory roles in plant response to stress, besides being involved in plant development and organogenesis. We analyzed the leaf miRNAome of two durum wheat cultivars (Cappelli and Ofanto) characterized by a contrasting water use efficiency, exposed to heat stress, and mild and severe drought stress. On the whole, we identified 98 miRNA highly similar to previously known miRNAs and grouped in 47 MIR families, as well as 85 novel candidate miRNA, putatively wheat specific. A total of 80 known and novel miRNA precursors were found differentially expressed between the two cultivars or modulated by stress and many of them showed a cultivar-specific expression profile. Interestingly, most in silico predicted targets of the miRNAs coming from the differentially expressed precursors have been experimentally linked in other species to mechanisms controlling stomatal movement, a finding in agreement with previous results showing that Cappelli has a lower stomatal conductance than Ofanto. Selected miRNAs were validated through a standardized and reliable stem-loop qRT-PCR procedure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Drought and high temperatures are among the most important environmental stresses affecting plant growth; consequently, the plants have evolved morphological and physiological adaptations, as well as molecular responses activated upon stress perception to cope with environmental constraints. The adaptation to heat and drought stress involves different strategies like shorter life cycle with accelerated flowering, reduction of water loss (i.e., stomatal closure, increased leaf cuticle thickness), and improvement of water uptake (i.e., deeper and/or larger root system). Morphological and physiological adaptations to environmental constraints are associated with wide transcriptional changes controlled by sophisticated molecular mechanisms (Cattivelli et al. 2008; Hu and Xiong 2014).
Durum wheat (Triticum turgidum subsp. durum) is an important cereal crop grown mainly in semi-arid environments, e.g., the Mediterranean regions, characterized by water scarcity and high temperatures. Consequently, drought and heat tolerance are main targets for durum breeding and many studies have been dedicated to the characterization of the physiological and molecular traits involved in the adaptation to these stress conditions. In this context, two durum wheat cultivars, Ofanto and Cappelli, have been extensively characterized since they represent genotypes with a significantly different response to drought (De Vita et al. 2007; Rizza et al. 2012; Panio et al. 2013; Aprile et al. 2013).
In previous studies, Cappelli consistently showed a constitutive higher water use efficiency (WUE), i.e. the ability of a crop to produce biomass per unit of water evapotranspired, compared to Ofanto, a finding associated with a lower stomatal conductance in Cappelli vs. Ofanto during all developmental stages and over a range of relative soil water contents (Rizza et al. 2012). Cappelli represents a typical isohydric–water saver genotype, whereas Ofanto can be considered an anisohydric–water spender cultivar (Levitt 1980; Tardieu and Simonneau 1998). Indeed, Cappelli closes stomata since on incipient drought, whereas the same response in Ofanto is activated after more severe drought conditions (Rizza et al. 2012). Aprile et al. (2013) reported that Ofanto activates a large set of well-known drought-related genes after drought treatment, while Cappelli shows a minimal modulation of gene expression in the same conditions associated to the constitutive expression of a set of genes that in Ofanto are induced only upon drought stress. These findings sustain the hypothesis that Cappelli activates the onset of drought mechanisms earlier then Ofanto, when water shortage is minimal.
The burst of deep sequencing technologies in the last few years has allowed the identification and quantification of many classes of small RNAs involved in gene regulation of different biological processes among them the abiotic stress response, revealing an additional level of posttranscriptional control of gene expression (Guerra et al. 2015). miRNAs, the best characterized small RNA category, are 20–24-nt-long single-stranded RNAs, originating from longer hairpin transcripts, transcribed from specific miRNA genes (MIR genes) by RNA polymerase II. miRNAs, acting upon sequence pairing as negative posttranscriptional regulators of gene expression, have emerged as important modulators in drought and heat tolerance and avoidance via control of drought and heat responsive genes (Rogers and Chen 2013; Budak et al 2015; Carrington and Ambros 2003; Ding et al. 2013). Substantial work has been carried out to investigate miRNA expression profiles and targets during drought stress in contrasting cultivars of several crop species like cowpea (Barrera-Figueroa et al. 2011), bread wheat (Ma et al. 2015), barley (Hackenberg et al. 2015; Ferdous et al. 2016), rice (Cheah et al. 2015) and durum wheat (Liu et al. 2015, 2016).
This work aims to study the modulation of the of the durum wheat miRNAome in response to two levels of drought stress and to heat stress in two durum wheat cultivars (Cappelli and Ofanto) characterized by contrasting WUE. A total of 80 precursor miRNAs (pre-miRNAs) were found differentially expressed in at least one comparison. Several drought or heat regulated pre-miRNAs were characterized by a genotype-specific expression profile and with predicted targets mostly involved in the control of stomatal opening according to the different level of WUE of the cultivars tested.
Materials and methods
Genetic materials and growth conditions
The experiment was performed using two durum wheat (Triticum turgidum subsp. durum) cultivars, Ofanto and Cappelli. The plants were grown at 20/18 °C day/night temperature with 16/8-h photoperiod and a relative soil water content (RSWC) maintained constantly at 85 %. Stress treatments were applied when plants reached the developmental stage of fully expanded mature third leaf. As for the drought stress, water was withheld until the RSWC dropped to 55 % (mild drought stress) or to 35 % (severe water stress), and leaves were harvested 2 and 3 days, respectively, after RSWC reached the desired threshold. Developmental stage and drought stress conditions were similar to those reported by Rizza et al. (2012) where a detailed analysis of WUE was carried out using the same cultivars employed in the current work. Leaves from control plants were harvested at the same time as leaves subjected to severe stress. Heat stress consisted in 3 h at 36 °C in presence of light, and leaves were harvested immediately after. Three biological replicates for each treatment were considered; in each replicate, all leaves of five plants were pooled together and used for RNA isolation.
RNA isolation and small RNA libraries preparation
Total RNA was extracted from wheat leaves tissues using TRIzol reagent (Invitrogen) with minor modifications. RNA quality and concentration were evaluated with the Agilent 2100 Bioanalyzer RNA 6000 Nano assay. All RNA samples were stored at −80 °C until further processing. The preparation of small RNA libraries was performed with the TruSeq Small RNA Sample Prep Kit (Illumina, San Diego, CA) according to the manufacturer’s instructions. Briefly, 1 μg of total RNA was ligated with two adapters at 3′ and 5′ ends. Adapter-ligated RNA was reverse-transcribed with SuperScript II Reverse Transcriptase (Invitrogen), then PCR-amplified (15 cycles). The cDNA libraries were purified on a 6 % TBE PAGE and quality and concentration were evaluated with the Agilent 2100 Bioanalyzer DNA1000 assay. Small RNA-seq data from 24 samples (12 from cv. Cappelli and 12 from cv. Ofanto—see Table 1) were performed using a 12-plex sequencing approach with 30-nt single-end reads on GAIIx sequencer (Illumina, San Diego, CA).
Bioinformatics prediction of known and novel miRNAs
Raw sequencing data were of good quality (mean sequence Phred quality score >30), and no quality filter was applied. Sequencing reads were trimmed using the program Cutadapt (Martin 2011) version 1.8.3 with the settings: --trim-n -a TGGAATTCTC.
For each sample, trimmed reads were mapped independently against the hexaploid Triticum aestivum cv. Chinese Spring reference genome version IWGSC2 downloaded from Ensembl Genomes (ftp://ftp.ensemblgenomes.org/pub/plants/release-26). Since the durum wheat genome is not yet available, only chromosomes belonging to the T. aestivum genomes A and B were considered as reference sequences to create a synthetic durum wheat reference. Mitochondrial and plastid genome were not considered in this analysis.
Bowtie (Langmead et al. 2009) version 1.0.1 was used to align trimmed reads to the reference genome allowing one mismatch. The mapping results of each sample were analyzed with ShortStack (Axtell 2013) version v. 2.0.9 with default settings. The initial annotation was refined by 183 high confident pre-miRNAs based on the current criteria for the annotation of plant miRNA (Meyers et al. 2008).
To identify conserved miRNAs, BLASTn (McGinnis and Madden 2004) with E-value <e−10 was applied using as subject all the hairpin sequences belonging to monocotyledonous species present in miRBase (Kozomara and Griffiths-Jones 2014) version 21. Results were manually inspected to verify the putative precursor sequences against the whole miRBase using the BLASTN function available on the website and to confirm that the most abundant duplex (5′ and 3′ miRNAs) was correctly extracted and named accordingly. Sequence abundance was normalized to Tag per one million (TP1M), to allow for libraries’ comparisons. Finally, homeologous miRNAs were identified based on sequence similarity along the entire stem-loop sequence using the clustering program CD-HIT (Fu et al. 2012) with sequence identity ≥0.95.
miRNA targets
The nonredundant set of 5′ and 3′ miRNA sequences was used to predict targets in the full set of cDNA transcripts annotated in the T. aestivum cv. Chinese Spring version IWGSC2 downloaded from Ensembl Genomes (ftp://ftp.ensemblgenomes.org/pub/plants/release-26). The program TargetFinder (http://github.com/carringtonlab/TargetFinder); Fahlgren et al. 2007; Fahlgren and Carrington 2010) with default parameters was applied, and only results with a score cutoff ≤4 were considered as putative miRNA targets.
Gene annotation have been obtained from URGI Sequence repository (http://wheat-urgi.versailles.inra.fr/Seq-Repository/Genes-annotations), taking the version 2.2 of gene models and the human readable description (file name: ta_IWGSC_MIPSv2.2_HighConf_HUMAN_READABLE_DESCS_2014Jul18.txt) which reports the functional descriptions for all high-confidence (HCS) gene models without splice variants.
Differential expression analysis
Differentially expressed pre-miRNAs were identified using the Bioconductor R package DESeq2 (Love et al. 2014) setting FDR ≤0.1 and using Wald hypothesis test. For each MIR locus, the total read counts mapped to the hairpin sequence in each condition was considered as the input dataset to perform the expression analysis. In total, seven pairwise comparisons were performed applying a FDR-adjusted p value ≤ 0.1: (i) Ofanto (all samples) vs. Cappelli (all samples), (ii) Cappelli DSI vs. Cappelli control, (iii) Cappelli DSII vs. Cappelli control, (iv) Ofanto DSI vs. Ofanto control, (v) Ofanto DSII vs. Ofanto control, (vi) Ofanto heat stress vs. control, and (vii) Cappelli heat stress vs. control.
Validation of miRNA expression profiles via stem-loop qRT-PCR
The expression profiles of miRNAs were assayed by stem-loop quantitative real-time PCR (qRT-PCR); the primers were designed according to Varkonyi-Gasic et al. (2007) and listed in Online Resource 1. The stem-loop reverse transcriptase primer for each miRNA consisted of a selfed stem-loop sequence (GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGAC) with the specificity conferred by a six nucleotide extension at the 3′ end, which is complementary to the last six nucleotides at the 3′ end of the miRNA target. The RT reactions were performed starting from 200 ng of total RNA, using Superscript III (Invitrogen) and carried out according to the manufacturer’s instructions. The reverse transcription products were amplified using a miRNA-specific forward primer and a universal reverse primer. The reactions were performed in triplicate on three independent biological replicates. The reactions were set up using SYBR Green PCR Master Mix (Life Technologies).
Different housekeeping genes, either durum wheat protein coding genes (polyubiquitin–Traes_7AL_5E8A63964 and OEP16–Traes_6AL_8262406D3.1) or small RNA nucleolar RNAs (snoRNAs), were considered for qPCR data normalization. snoRNAs sequences were taken from plant snoRNA online database (http://bioinf.scri.sari.ac.uk/cgi-bin/plant_snorna/home), and primers were designed on the loop region (carefully avoiding regions partly on the loop and partly on the stem—Online Resource 1). Ta-snoR10 showed an expression level similar to mid-expressed miRNAs and a good stability in all the conditions of the present experiment and was therefore chosen as endogenous control.
Results
Libraries statistics and analyses
A total of 24 libraries were constructed from RNA isolated from leaves of two durum wheat cultivars (Cappelli and Ofanto), subjected to two levels of drought (mild and severe) or to heat stress (Table 1). The libraries were deep sequenced with Illumina technology, and a total of 74,277,547 raw reads were generated. Out of them 59,974,281 clean reads were mapped on the A and B genomes of bread wheat considered as the putative durum wheat genome (Online Resource 2).
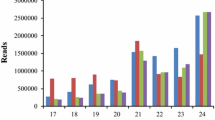
Small RNA size distribution for each library revealed a typical 2-peaks profile, as expected for Dicer-like protein (DCL) products (Fig. 1). Most of the libraries show the 24-nt peak higher than the 21-nt peak, as already observed for leaf-derived libraries in monocots (Ma et al. 2015; Xu et al. 2014; Wu et al. 2016; Bertolini et al. 2013), with the exception of the library from Cappelli in control conditions (CC) and of the two libraries from heat stressed plants (CHS and OHS), that show a 24-nt peak as high as the 22-nt peak. These libraries have a reduced proportion of siRNAs (typically 24 nt long), and a significant shift toward 21–22-nt molecules (typically miRNAs). miRNAs with a length of 22 nt facilitate the triggering of secondary siRNA production from their target transcripts, enhancing their silencing power (Cuperus et al. 2010; Chen et al. 2010).

Small RNA size distribution of durum wheat libraries. X-axis: length in nucleotides (nt); Y-axis: raw abundances for each size class as an average of three biological replicates, in Cappelli (upper panel) and in Ofanto (lower panel). CC Cappelli control, CDSI Cappelli drought stress I, CDSII Cappelli drought stress II, CHS Cappelli heat stress, OC Ofanto control, ODSI Ofanto drought stress I, ODSII Ofanto drought stress II, OHS Ofanto heat stress
Identification of durum wheat miRNA
The public miRNA repository miRBase (www.mirbase.org v.21) contains only one durum wheat miRNA, and few works have been published on this species (Liu et al. 2015). To advance the knowledge on miRNAs in this crop, the reads mapped to the A and B genomes of bread wheat, considered as the putative durum wheat genome, were analyzed with ShortStack pipeline (Axtell 2013) annotating a total of 37,933 clusters corresponding to genomic regions of small RNA accumulation. Successive filtering steps identified 183 high confident miRNA, matching the criteria for miRNA annotation (Meyers et al. 2008), each producing one or more miRNA duplexes. BLASTn search against miRBase database and manual refinement of in silico produced data, identified a total of 98 miRNA homologous to previously identified plants miRNAs and 85 novel miRNAs, putatively wheat specific, described hereafter as known and novel miRNAs, respectively (Online Resource 3; Fig. 2).

Pre-miRNA secondary structure as calculated by RNAfold. Stem-loop secondary structures of five unknown miRNAs tested for by qRT-PCR (qRT-PCR data shown in Fig. 6). The color code indicates the abundance of the sequencing tag mapping to each position
The set of 98 known miRNA correspond to 47 different MIR families, including 16 out of 21 highly conserved families, shown to be largely represented in almost all plant species (Cuperus et al. 2011). MIR169 (13 members), MIR156, and MIR167 (8 members) were the largest families, whereas no miRNAs were annotated as MIR408, MIR394, MIR395, MIR162, and MIR168. We annotated as 5′ and 3′ miRNAs the most abundant duplex produced by each precursor, even if sometimes it was not the most similar to previously annotated miRNAs. This is the case, for example, of Cluster_37565 and Cluster_37589, members of MIR169 family, whose most expressed miRNAs was 5 nt shifted when compared to the most canonical annotated miR169, and of Cluster_37107, whose most expressed miRNA was 2 nt shifted in respect to the annotated hvu_miR5048. In some cases, we identified the precursor as belonging to a known MIR family, even if the produced miRNA duplex is not similar with any annotated miRNA: this is the case of Cluster_16383 (miR1128) and Cluster_12532 (miR9781) that were longer than their homologous precursors, producing the most abundant duplex upstream and downstream the homology region. It should be noticed, however, that some known monocots specific families, recently identified, could have been mis-annotated. For instance, miR1122, miR1127, miR1128, miR1135, and miR1136, all belonging to the MIR1122 gene family, have been often identified only in silico, and when NGS data are available, they do not confirm previous annotations. It is not surprising, then, if the present data are not always in agreement with deposited sequences for these families.
We also identified some precursors producing phased-miRNAs, i.e., miRNAs excised from the stem of the precursor, in phase with each other (Zhang et al. 2010; Bologna et al. 2009; Addo-Quaye et al. 2009; Kurihara and Watanabe 2004). miR159 and miR169 have been already identified as miRNAs typically producing phased miRNAs (Belli Kullan et al. 2015; Contreras-Cubas et al. 2012), and here, we show three pre-miR169 producing two different duplexes (Fig. 3). ShortStack did not identified phased miRNAs for miR159 (Cluster_12004) as they were not perfect duplexes, but their abundance and the evolutionary conservation with other species sustain their existence.

MIR169 and MIR159 loci generating more than one duplex. Sequence and dot-bracket notation representing RNA stem-loop structure of some MIR loci generating more than one distinct duplex from the same precursor. For each precursor, name, MIR family, and mapping position (chromosome, coordinates, strand) are presented. Below the dot-bracket notation, distinct miRNA duplexes are aligned and the raw abundance (a) and length (l) of the reads are shown
Among the 85 novel miRNAs, potential candidates were grouped based on the similarity of miRNA sequences into 69 gene families, of which only 10 have more than one member.
As expected, the abundance of pre-miRNAs, summing up all the 24 libraries, is different between known and novel ones, with known pre-miRNAs far more expressed with an average expression of 236,655 TP1M against 9503 TP1M of the newly identified ones (Online Resource 4). The most expressed pre-miRNAs were Cluster_27773 (miR166), Cluster_29830 and Cluster_32083 (two members of MIR156 family), and Cluster_12004 (miR159), well-known and conserved MIR families, and Cluster_37107 (miR5048) and Cluster_24041 (miR5168), belonging to two monocots specific families, not yet well characterized.
Among unknown miRNAs, Cluster_29152 and Cluster_19417 were the most abundant precursors in the sequenced libraries with 172,629 TP1M and 136,609 TP1M, respectively. Cluster_29152, together with Cluster_4530, Cluster_8217, Cluster_27953, Cluster_13866, and Cluster_15398 are similar to miR531, although this similarity is not sufficient to define these miRNAs as putative members of this family. We also identified some miRNAs whose precursor and mature sequences were similar to two different MIR families (usually low confidence or uncharacterized MIR families). We annotated these miRNAs as novel, as their homology with already known miRNAs is not well defined. Moreover, Cluster_19417 shows a pattern of phased-small RNA production, although the phasing is not perfect and ShortStack did not select the other duplexes.
Cappelli and Ofanto were characterized by a different miRNA population even in absence of stress. Figure 4 shows the results of the comparison between the control sample of the two cultivars for those pre-miRNAs with at least a 1.5 ratio between Cappelli and Ofanto, considering the normalized abundances and summing up precursors’ abundance of each MIR family (Online Resource 4). The most striking examples are Cluster_35513 and MIR5200 family nearly 15- and 10-fold, respectively, more expressed in Cappelli, while Cluster_29152 and Cluster_30941 were 80- and 13-fold, respectively, more expressed in Ofanto. Within novel miRNAs, Cluster_8217 and Cluster_15398 were undetectable in Cappelli but clearly expressed in Ofanto (about 1900 and 1300 TP1M, respectively). This analysis suggests that these novel pre-miRNAs, putatively species specific, could have an important role in varietal differences.

Profile of pre-miRNA abundance in Ofanto and Cappelli. pre-miRNA loci normalized (TP1M) abundance in the small RNA libraries from Ofanto (dark blue) and Cappelli (light green) in control conditions have been averaged and summed up for known MIR families. Only pre-miRNA showing at least 1.5 ratio in the Ofanto vs. Cappelli comparison are shown. Pre-miRNAs have been divided in the three panels based on their abundances, to appreciate differences also among low expressed loci
Genomic organization of durum wheat MIR genes
We observed an even distribution of the identified MIR loci in the A and B genomes of bread wheat considered as the putative durum wheat genome (Fig. 5), with the exception of chromosome 3B, the largest one that hosts a relative low number of predicted miRNAs (16 loci in more than 750 Mbases). Very few miRNAs are organized in tandem repeats (Online Resource 3). The most striking examples are the three members of MIR5200 family, clustered together in a 1.5-kb region on chromosome 7B. Additionally, we found the two members of MIR1432 family in 3.5 kb on chromosome 2A, two miR9674 in 1.5 kb on chromosome 4B, and two miR167 in nearly 3 kb on chromosome 5B. Two unknown and novel miRNAs are also clustered together, and based on their mature sequence, they do constitute a MIR family (Cluster_34228 and Cluster_34229, in 2 kb on chromosome 7A).

Chromosome distribution of MIR loci in the Triticum durum synthetic genome. MIR loci, shown as black bars, are plotted on the circular representation of the A and B genomes of bread wheat considered as the putative durum wheat genome, based on the coordinates of the predicted pre-miRNAs
Considering the tetraploid nature of the genome of durum wheat, a specific search was dedicated to the identification of the putative homeologous miRNAs, defined as miRNA sharing more than 95 % of identity in their precursor sequences and sitting on homeologous chromosomes. We found eight couples of homeologous miRNAs: three on chromosomes 5A-5B (Cluster_24561 and Cluster_27799; Cluster_24041 and Cluster_26880; Cluster_23928 and Cluster_26932), two on chromosomes 2A-2B (Cluster_7528 and Cluster_11139; Cluster_10594 and Cluster_6194), Cluster_19682 and Cluster_21808 on chromosomes 4A-4B, Cluster_29830 and Cluster_32083 on chromosomes 6A-6B, and Cluster_34415 with Cluster_36805 on chromosomes 7A-7B.
In silico identification of target genes
We predicted the putative targets for the miRNA sequences here identified based on sequence similarity and using the program TargetFinder. Although in silico approaches can still produce large numbers of false-positive predictions, these tools are useful to describe a tentative role of newly identified and previously unknown miRNAs. When the complete set of miRNAs identified in the current work, comprising a total of 324 nonredundant miRNA sequences (considering 5′ and 3′ sequences) was used as input, a total of 257 miRNAs yielded at least one target, for a total of 2252 different targets (Online Resource 5). Conserved miRNAs usually share the same targets across different plant species (Chen 2009); indeed, most target predictions for highly conserved miRNAs are as expected from predictions and validations in other species.
pre-miRNAs differentially expressed between Ofanto and Cappelli in control and drought stress conditions
The DESeq2 R/Bioconductor package (Love et al. 2014) was used to gain statistical evidence for differential expression of known and novel pre-miRNAs between Ofanto and Cappelli grown in control condition or exposed to drought stress (DSI and DSII). A total of 80 pre-miRNAs were found to be differentially regulated in at least one comparison.
The comparison Ofanto vs. Cappelli yielded a total of 44 pre-miRNAs (25 known and 19 novel) as differentially regulated, suggesting a constitutively different expression of these pre-miRNAs in the two genotypes (Table 2). Some pre-miRNAs were found more expressed in Cappelli, e.g., miR827 (Cluster_10488), miR171 (Cluster_7479), and miR5200 (Cluster_35769 and Cluster_35770), while others were more expressed in Ofanto, e.g., miR156 (Cluster_12326), miR167 (Cluster_18670), Cluster_15398, and Cluster_29152. Interestingly, the target genes of some of the miRNAs originating from these pre-miRNAs are involved in the regulation of stomatal movements. For example, the predicted target of miR5200 (Cluster_35769 and Cluster_35770) is the highly conserved florigen gene FLOWERING LOCUS T (FT), a gene also involved in the control of stomatal movement (Wu et al. 2013).
GRAS (GIBBERELLIC–ACID INSENSITIVE (GAI), REPRESSOR of GAI (RGA), SCARECROW (SCR)) proteins are plant-specific proteins with important roles in plant growth, development, phytohormone signal transduction, and stress response (Bolle 2004; Xu et al. 2015). We found GRAS proteins as predicted targets for miR171 from Cluster_7479, a miRNA constitutively more expressed in Cappelli compared to Ofanto (Table 2). The target of miR156 from Cluster_12326, constitutively more expressed in Ofanto vs. Cappelli, is the conserved SQUAMOSA PROMOTER BINDING PROTEIN LIKE-PROTEIN (SBP) transcription factor, which is known to be fundamental for leaf growth and development (Wu and Poethig 2006).
To identify drought-associated pre-miRNAs in Cappelli and Ofanto plants exposed to a mild (DSI) and to a severe (DSII) drought stress have been compared with plants grown under optimal water conditions. A total of 11 (9 known and 2 novel) and 15 pre-miRNAs (12 known and 3 novel) were found differentially expressed in Cappelli, while a total of 10 (9 known and 1 novel) and 16 pre-miRNAs (10 known and 6 novel) were found differentially expressed in Ofanto during mild and severe stress, respectively. These drought-related pre-miRNAs include sequences with the same expression profile in the two cultivars as well as sequences with a cultivar-specific expression profile.
The repression of miR169 in response to drought has been reported for several species (Khraiwesh et al. 2012; Sunkar et al. 2012; Ding et al. 2013) with a putative function in the control of stomatal movement. A strong downregulation of five family members of MIR169 has been detected in both Cappelli and Ofanto with some differences concerning their fold change and the time course expression profiles: Cluster_11103 showed a repression trend, visible already during the mild stress (DSI) in both genotypes. Cluster_21808 showed the same trend in both genotypes but higher log2 fold change in Cappelli in both DSI and DSII conditions. One of the predicted target of miR169 (Cluster_21808) is GLUTAMATE DECARBOXYLASE (GAD), an enzyme involved in the GABA biosynthesis, whose depletion affects stomatal closure and drought tolerance in Arabidopsis (Mekonnen et al. 2016). On the contrary, Cluster_10976 was downregulated earlier in Cappelli, while in Ofanto, was downregulated in DSII only. The predicted targets of miR169 (Cluster_10976) are different subunits of NUCLEAR FACTOR Y, SUBUNIT A (NFYA), a plant transcription factor with important roles in development and response to environmental stresses (Kumimoto et al. 2008). Another member of the same MIR family, Cluster_8221, was constitutively downregulated in Cappelli vs. Ofanto, while Cluster_28781 was strongly downregulated in Ofanto only in DSI and the predict targets are different subunit of NFYA. Similarly, the novel miRNA Cluster_36996 displayed a downregulation already during mild stress conditions in both genotypes with a higher log2 fold change in Cappelli than in Ofanto. Interestingly, a possible target of this candidate miRNA is NFYA exactly the same target of miRNA169 (Cluster_10976).
MIR159 represents another drought-related miRNA family involved in the ABA mediated pathways, and the corresponding targets are MYB transcription factors, positive regulators of ABA signalling (Reyes and Chua 2007). Ofanto and Cappelli showed an upregulation of Cluster_34804 (MIR159) in severe drought stress (DSII) with a log2 fold change ≥3. Likewise, Cluster_10488 and Cluster_7304 (MIR827) and the novel miRNA Cluster_29152 showed the same expression profile with a significant upregulation in DSII in both cultivars.
Among the pre-miRNAs with cultivar-specific expression profile, in Cappelli, we found, among others, miR5200 (Cluster_35769 and Cluster_35770), miR393 (Cluster_14246), miR171 (Cluster_23199), miR5048 (Cluster_ 37107), and the novel Cluster_21463 all induced, while miR399 (Cluster_22915), miR528 (Cluster_23249), and miR1136 (Cluster_20013) were repressed in response to drought stress. On the other hand, in Ofanto, miR166 (Cluster_27773) and Cluster_29324 were upregulated, while miR1432 (Cluster_6638) and Cluster_33043 were repressed. Among these, miR1432 (Cluster_6638) showed a substantial decrease in Ofanto DSII only; its downregulation under drought stress was described also in wild emmer, the ancestor of durum wheat (Kantar et al. 2011). miR1432 (Cluster_6638) is predicted to target CALMODULIN LIKE-43, suggesting its involvement in calcium signalling. This network is implicated in the response to abiotic stress and Ca2+ signals are core regulators of stomatal aperture (Dodd et al. 2010; Batistič and Kudla 2012).
Distinct members of MIR167 family, known to be regulated by drought stress also in other plant species (Ren et al. 2012; Phookaew et al. 2014), showed a peculiar expression profile in Cappelli and Ofanto. mR167 (Cluster_ 26272) was induced in both mild and severe drought stress in Cappelli only, while miR167 (Cluster_ 28193) was induced in Cappelli DSII only and miR167 (Cluster_ 22968) was upregulated after severe stress only in Ofanto. The predicted target gene of miR167 (Cluster_26272, Cluster_28193) is ATPase FAMILY GENE 2 PROTEIN that has been suggested to be also involved in stomatal control (Wang et al. 2013).
miRNAs differentially expressed in Ofanto and Cappelli during heat stress
The analysis of the pre-miRNAs differentially expressed in response to heat treatment have yielded a total of 12 (6 known and 6 novel) and 25 (18 known and 7 novel) pre-miRNAs in Cappelli and Ofanto, respectively (Table 2). The expression profile showed a common response for different members of MIR169 family upregulated in both genotypes, but miR169 (Cluster_8221) which is downregulated in Cappelli only. Similarly, two novel pre-miRNAs (Cluster_ 36747 and Cluster_ 36996) were downregulated after heat stress in both genotypes.
The members of MIR167 family showed a different behavior in two cultivars exposed to heat stress. The upregulation of miR167 (Cluster_ 26272) was detected in Cappelli, a trend already seen in the same cultivar subjected to drought stress. On the other hand, the downregulation of miR167 (Cluster_26273), miR167 (Cluster_32198), and miR167 (Cluster_ 2296818670) in response to heat treatment was found in Ofanto only. Overall, Ofanto downregulated more miRNA genes in response to heat stress than Cappelli, e.g., miR159 (Cluster_34804), miR166 (Cluster_36963), miR167 (Cluster_26273), miR167 (Cluster_18670), miR167 (Cluster_26272), miR9776 (Cluster_11139), miR5200 (Cluster_35769 and Cluster_35770), miR9781 (Cluster_28123), and miR159 (Cluster_34804).
Validation of differentially expressed miRNAs by stem-loop qRT-PCR
Stem-loop qRT-PCR was carried out to validate the expression profiles of ten (six known and four novel) miRNAs identified as differentially expressed after deep sequencing and selected on the basis of their expression level and the biological relevance of their putative targets (miR156/Cluster_12326, miR167/Cluster_18670, miR1432/Cluster_6638, miR159/Cluster_34804, miR169/Cluster_11103, miR169/Cluster_8221, Cluster_29324, Cluster_34270, Cluster_36747, Cluster_36996). Log2 fold changes of selected miRNAs are shown in Fig. 6. qRT-PCR produced reliable data for seven miRNAs; for these genes, the qRT-PCR analysis showed the same expression profile across all samples as Illumina deep sequence analysis. qRT-PCR analysis of the remaining three miRNAs showed few inconsistencies (one each for miR169/Cluster_11103 and Cluster_36996, and two for miR169/Cluster_8221) when compared to Illumina data (Fig. 6). Overall, the qRT-PCR analysis validates the deep sequencing data with only 4 inconsistencies over 60 comparisons.

qRT-PCR expression analysis of selected stress responsive miRNAs. Log2 fold change (FC) indicates the miRNA expression in the different comparisons: Cappelli drought stress I (CDSI) vs. Cappelli control (CC); Cappelli drought stress II (CDSII) vs. CC; Cappelli heat stress (CHS) vs. CC; Ofanto drought stress I (ODSI) vs. Ofanto control (OC); Ofanto drought stress II (ODSII) vs. OC; Ofanto heat stress (OHS) vs. OC. The wheat snoR10 was used as an endogenous control. Asterisk indicates the samples with different expression trend between Illumina deep sequence and qRT-PCR data
Discussion
We analyzed the leaf miRNAome of two durum wheat cultivars characterized by a different water use efficiency (Rizza et al. 2012), exposed to two levels of drought stress and to heat stress. On the whole, we identified 98 miRNAs highly similar to previously known miRNAs and grouped in 47 MIR families, as well as 85 novel candidate miRNA, putatively wheat specific. We found some minor variations in length or nucleotidic sequence in respect to known monocots miRNAs for most predictions, with the exception of few miR169 precursors and members of MIR9781 and MIR1122 families, which could have been mis-annotated in T. aestivum.
Notably, we did not identified five highly conserved MIR families. MIR408 and MIR162 did not have almost any sequencing tags, suggesting their absence in the leaf of young durum wheat plants. A similar result was reported for miR162 in Brachypodium young leaves in a previous work (Bertolini et al. 2013). MIR394, MIR395, and MIR168 were present in our libraries, but were unable to pass the stringent parameters of the ShortStack pipeline.
Considering the 85 novel miRNAs identified, they are, on average, less expressed than known miRNAs, as expected, and rarely grouped into families. The main exception is the case of the six novel miRNAs similar to MIR531 family (Cluster_29152, Cluster_4530, Cluster_8217, Cluster_27953, Cluster_13866, Cluster_15398), but not enough to be considered real members of this family.
Ofanto and Cappelli, two durum wheat cultivars with different miRNAomes
This work is dedicated to the analysis of two durum wheat cultivars well characterized at physiological level for their different WUE, largely due to the different stomatal conductance (Rizza et al. 2012). It is known that plants may have different physiological behaviors with respect to water fluxes with important consequences for their survival, growth, and yield. Cappelli, a typical old cultivar, displays a water conserving behavior (isohydric plant); on the contrary, a modern cultivar such as Ofanto, displays a risk-taking behavior (anisohydric plants). Isohydric plants maintain a relatively constant leaf relative water content by reducing evaporation when facing with water stress. Anisohydric plants allow leaf water potential to decrease with rising evaporative demand, reaching a lower relative water content under drought conditions compared to situations in which they are well-watered (Tardieu and Simonneau 1998). Stomatal control is the key for the regulation of evapotraspiration; nevertheless, there is limited knowledge of the molecular and cellular criteria differentiating these two types of plants, which constrains our ability to manipulate the stomata behavior of crop species to improve either water use efficiency or drought tolerance (Moshelion et al. 2015).
We initially analyzed the varietal difference between Ofanto and Cappelli comparing the relative pre-miRNA expression. Differential expression levels are evident for genes such as Cluster_35770 and Cluster_35769 (MIR5200) far more expressed in Cappelli than in Ofanto, a precursor whose mature has FT as predicted target. Similarly, many novel pre-miRNAs were also strongly modulated in Cappelli vs. Ofanto or were shut down in one of the two cultivars (Table 2, Fig. 4). This is the case, for example of Cluster_15398 and Cluster_29152, more expressed in Ofanto, and of Cluster_35513 and Cluster_24466 much more expressed in Cappelli. This particular feature suggests that novel and putatively wheat specific miRNAs are involved in the definition of varietal differences. A deeper understanding of the functional role of such miRNAs is instrumental to further characterize the small RNA–driven differences between these two contrasting genotypes.
A comparison of the miRNAs identified as responsive to drought in durum wheat leaves with those described in response to the same stress in durum wheat and wild emmer root tissues identified only two miRNAs with a similar expression profile (miR169 and miR1136, Akpinar et al. 2015), suggesting that variation in the miRNAome in response to stress are largely tissue specific. Notably, the miR1136 in both experiments has been found downregulated only in the tolerant genotypes.
A total of 49 pre-miRNAs (known and novel) were found differentially expressed in Ofanto (37) and Cappelli (24) during drought and heat stresses. Some of them were part of a common response to stress, while for most of them, we have highlighted a cultivar specific expression profiles. A strong upregulation of miR159 in response to drought is known in the literature (Reyes and Chua 2007), and it was confirmed in both cultivars. This miRNA targets, in durum wheat also, MYB33 and MYB65, two transcription factors of the ABA signalling pathway. miR159 overexpression in Arabidopsis suppresses MYB33 transcript level making plants hyposensitive to ABA and desensitize hormone signalling during seedling stress responses (Reyes and Chua 2007).
Our work highlights that many of the differences in miRNA expression between the two durum wheat cultivars with contrasting stomatal conductance and exposed to drought stress have as putative target genes that in other species have been experimentally linked to stomatal controlling mechanisms. MIR169 is one of the largest and conserved MIR families; its involvement in drought stress is reported for several species although with contrasting expression patterns. For example, two members of the MIR169 family in rice, miR169g and miR169n/o, and one member in tomato, miR169c, were upregulated by drought stress. In addition, overexpression of miR169c in tomato reduced stomatal conductance and water loss showing enhanced drought tolerance in transgenic tomato lines (Zhang et al. 2011; Zhao et al. 2009). Conversely, the repression of MIR169 family members has been reported for several species and also associated to drought stress response (Khraiwesh et al. 20122012; Sunkar et al. 2012; Ding et al. 2013). In Arabidopsis, the downregulation of miR169a, which targets the NFYA5, contributes to the high level of NFYA5 expression detected in response to drought and ABA (Kumimoto et al. 2008). NFYA5 was found highly expressed in vascular tissues and guard cells, and the analysis of nfya5 knockout plants and of miR169a or NFYA5 overexpression lines showed that NFYA5 has a role in the stomatal control and, hence, drought resistance (Li et al. 2008). In durum wheat, we found a downregulation, more in Cappelli than in Ofanto, of some members of MIR169 family having NFYA5 has putative target. Furthermore, the novel miRNA Cluster_36996, targeting the same NFYA as miR169 (Cluster_10976), follows the same expression pattern as miR169 being more downregulated in Cappelli than in Ofanto in response to drought. One of the predicted targets of miR169 (Cluster_21808), more downregulated in Cappelli than in Ofanto, is glutamate decarboxylase (GAD), a key enzyme of the gamma-aminobutyric acid (GABA) biosynthesis. A rapid accumulation of GABA during abiotic and biotic stresses is well documented. Mekonnen and collaborators (2016) found that gad1/2 mutant of Arabidopsis wilted earlier than wild type when exposed to drought stress due to increased stomatal aperture and to a deficit in stomatal closure, driven by a reduced GABA accumulation.
One of the predicted targets of the novel Cluster_15398, constitutively more expressed in Ofanto than in Cappelli, is an ATP-BINDING CASSETTE (ABC) TRANSPORTER B FAMILY member. ABC proteins are key players of cellular processes involved in auxin transport, lipid catabolism, xenobiotic detoxification, disease resistance, and stomatal function (Cho and Cho 2013). Interestingly, ABCB14 has been known as a malate importer modulating stomatal movement in guard cells (Lee et al. 2008).
miR5200 (Cluster_35769 and Cluster_35770), constitutively more expressed in Cappelli and further upregulated under severe drought stress in the same cultivar, targets FT. Evidences in the literature demonstrated that miR5200 directly mediate FT post transcriptional modulation in Pooideae plants (Wu et al. 2013), and it is known that FT is expressed in guard cells and regulates stomatal opening. Accordingly, transgenic plants overexpressing FT in guard cell showed open stomata, while a loss of function FT allele exhibited closed stomata (Kinoshita et al. 2011).
miR393 is specifically upregulated in Cappelli exposed to severe drought stress. Transgenic overexpression of miR393 in rice resulted in hyposensitivity to synthetic auxin analogue treatments (Xia et al. 2012), suggesting that miR393 may regulate auxin signalling and would thus reduce plant growth under drought stress. miR393 was found to target TRANSPORT INHIBITOR RESPONSE 1 (TIR1), known as an auxin receptor and positive regulator of auxin signaling that acts via degradation of Aux/IAA proteins (Dharmasiri and Estelle 2002; Windels and Vazquez 2011).
Additional pre-miRNAs with putative targets that in other species have been experimentally linked to stomatal regulation are miR1432 (Cluster_6638) and miR167. The first one (downregulated in Ofanto), has CALMODULIN LIKE-43 as putative target, a gene known for its involvement in the response of stomata to ABA (De Silva et al. 1985). miR167 (Cluster_26272, Cluster_28193), a sequence upregulated in Cappelli in response to drought treatments, has an ATPASE FAMILY GENE 2 PROTEIN as putative target. H + -ATPases are known to be responsible for the hyperpolarization of the plasma membranes and their inhibition is essential to induce ABA mediated stomatal closure (Merlot et al. 2007). Blue light activated H + -ATPase induces hyperpolarization of the plasma membrane which allows K+ uptake which, in turn, leads the swelling of guard cells and stomatal opening. Noteworthy, FT is suggested to be a positive regulator for stomatal opening via its effect on the activation status of the plasma membrane H + -ATPase (Kinoshita et al. 2011). Overexpression of plasma membrane H + -ATPase in guard cells promotes light-induced stomatal opening and enhances plant growth (Wang et al. 2013).
The analysis of the pre-miRNAs differentially expressed in response to heat treatment has led to 12 and 25 miRNA in Cappelli and Ofanto, respectively. Noteworthy, most of the miRNAs modulated in Ofanto were downregulated (15 out of 25), while in Cappelli, only three miRNAs showed the same expression trend. miR159 is the best known heat-responsive miRNA. Its putative target genes were identified as MYB domain protein (MYB33, MYB65). OsMYB55 can enhance the vegetative growth and improve grain yield of rice growth under high temperature conditions increasing the level of total amino acids (glutamine acid, proline, arginine, and GABA) (El-kereamy et al. 2012). miR159 was also shown to regulate TaGAMYB in bread wheat and rice transgenic lines overexpressing wheat precursor of miR159 showed a loss of heat tolerance suggesting its involvement in a heat stress-related signaling pathway. Interestingly, the TaGAMYB1 homologous Arabidopsis genes (AtMYB33 and AtMYB65) are also heat inducible (Wang et al. 2012). In the Cappelli-Ofanto experimental system, we confirmed the downregulation of miR159 (Cluster_34804) during heat treatment in Ofanto only, a finding that might suggest a minor heat sensitivity in this modern cultivar.
Transcriptomic analyses have shown that Ofanto activates a large set of genes in response to drought and heat stress, while Cappelli is characterized by the constitutive expression of several stress-related genes that in Ofanto are regulated only upon stress treatment (Aprile et al. 2013). This trend is confirmed also under the miRNA perspective, in particular during the heat stress response.
Overall, the data here presented indicate that Cappelli and Ofanto are characterized by significantly different miRNAomes and many of the differences in pre-miRNA expression in response to drought stress can be putatively linked to the stomatal controlling mechanisms. This finding matches the different stomatal conductance that was found when the same cultivars were investigated at physiological level (Rizza et al. 2012). Taken together, these studies suggest that the old cultivar Cappelli has a more conservative behavior with several stress-related mechanisms constitutively activated. On the contrary, the modern cultivar Ofanto represents a typical risk-taken plant and under conditions characterized by adequate irrigation and mild to moderate abiotic stress, this strategy proves advantageous, and risk-taken plants may outperform isohydric plants in terms of growth and yield (Moshelion et al. 2015).
References
Addo-Quaye C, Snyder JA, Park YB, Li YF, Sunkar R, Axtell MJ (2009) Sliced microRNA targets and precise loop-first processing of MIR319 hairpins revealed by analysis of the Physcomitrella patens degradome. RNA 15:2112–2121. doi:10.1261/rna.1774909
Akpinar BA, Kantar M, Budak H (2015) Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Funct Integr Genomics 15:587–598. doi:10.1007/s10142-015-0453-0
Aprile A, Havlickova L, Panna R, Marè C, Borrelli GM, Marone D, Perrotta C, Rampino P, De Bellis L, Curn V, Mastrangelo AM, Rizza F, Cattivelli L (2013) Different stress responsive strategies to drought and heat in two durum wheat cultivars with contrasting water use efficiency. BMC Genomics 14:821
Axtell MJ (2013) ShortStack: comprehensive annotation and quantification of small RNA genes. RNA 19:740–751. doi:10.1261/rna.035279.112
Barrera-Figueroa BE, Gao L, Diop NN, Wu Z, Ehlers JD, Roberts PA, Close TJ, Zhu J, Liu R (2011) Identification and comparative analysis of drought-associated microRNAs in two cowpea genotypes. BMC Plant Biol 11:127
Batistič O, Kudla J (2012) Analysis of calcium signaling pathways in plants. Biochim et Biophys Acta 1820(8):1283–1293
Belli Kullan J, Lopes Paim Pinto D, Bertolini E, Fasoli M, Zenoni S, Tornielli GB, Pezzotti M, Meyers BC, Farina L, Pè ME, Mica E (2015) miRVine: a microRNA expression atlas of grapevine based on small RNA sequencing. BMC Genomics 16:393. doi:10.1186/s12864-015-1610-5
Bertolini E, Verelst W, Horner DS, Gianfranceschi L, Piccolo V, Inzé D, Pè ME, Mica E (2013) Addressing the role of microRNAs in reprogramming leaf growth during drought stress in Brachypodium distachyon. Mol Plant 6:423–443. doi:10.1093/mp/sss160
Bolle C (2004) The role of GRAS proteins in plant signal transduction and development. Planta 218:683–692
Bologna NG, Mateos JL, Bresso EG, Palatnik JF (2009) A loop-to-base processing mechanism underlies the biogenesis of plant microRNAs miR319 and miR159. EMBO J 28:3646–3656. doi:10.1038/emboj.2009.292
Budak H, Kantar M, Bulut R, Akpinar BA (2015) Stress responsive miRNAs and isomiRs in cereals. Plant Sci 235:1–13
Carrington JC, Ambros V (2003) Role of microRNAs in plant and animal development. Science 301:336–338
Cattivelli L, Rizza F, Badeck FW, Mazzucotelli E, Mastrangelo AM, Francia E, Marè C, Tondelli A, Stanca AM (2008) Drought tolerance improvement in crop plants: an integrated view from breeding to genomics. Field Crop Res 105:1–14
Cheah BH, Nadaraja K, Divate MD, Wickneswari R (2015) Identification of four functionally important microRNA families with contrasting differential expression profile between drought-tolerant and susceptible rice leaf at vegetative stage. BMC Genomics 16(1):692
Chen X (2009) Small RNAs and their roles in plant development. Annu Rev Cell Dev Bi 25:21–44. doi:10.1146/annurev.cellbio.042308.113417
Chen HM, Chen LT, Patel K, Li YH, Baulcombe DC, Wu SH (2010) 22-Nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc Natl Acad Sci U S A 107:15269–15274. doi:10.1073/pnas.1001738107
Cho M, Cho HT (2013) The function of ABCB transporter in auxin transport. Plant Signal Behav 8(2), e22990
Contreras-Cubas C, Rabanal FA, Arenas-Huertero C, Ortiz MA, Covarrubias AA, Reyes JL (2012) The Phaseolus vulgaris miR159a precursor encodes a second differentially expressed microRNA. Plant Mol Biol 80:103–115. doi:10.1007/s11103-011-9847-0
Cuperus JT, Carbonell A, Fahlgren N, Garcia-Ruiz H, Burke RT, Takeda A, Sullivan CM, Gilbert SD, Montgomery TA, Carrington JC (2010) Unique functionality of 22-nt miRNAs in triggering RDR6-dependent siRNA biogenesis from target transcripts in Arabidopsis. Nat Struct Mol Biol 17:997–1003. doi:10.1038/nsmb.1866
Cuperus JT, Fahlgren N, Carrington JC (2011) Evolution and functional diversification of MIRNA genes. Plant Cell 23:431–442. doi:10.1105/tpc.110.082784
De Silva DLR, Cox RC, Hetherington AM, Mansfield TA (1985) Suggested involvement of calcium and calmodulin in the responses of stomata to abscisic acid. New Phytol 101:555–563
De Vita P, Li Destri Nicosia O, Nigro F, Platani C, Riefolo C, Di Fonzo N, Cattivelli L (2007) Breeding progress in morpho-physiological, agronomical and qualitative traits of durum wheat cultivars released in Italy during the 20th century. Eur J Agron 26:39–53
Dharmasiri S, Estelle M (2002) The role of regulated protein degradation in auxin response. Plant Mol Biol 49(3-4):401–409
Ding Y, Tao Y, Zhu C (2013) Emerging roles of microRNAs in the mediation of drought stress response in plants. J Exp Bot 64:3077–3086. doi:10.1093/jxb/ert164
Dodd AN, Kudla J, Sanders D (2010) The language of calcium signaling. Annu Rev Plant Biol 61:593–620
El-kereamy A, Bi YM, Ranathunge K, Beatty PH, Good AG, Rothstein LG (2012) The rice R2R3-MYB transcription factor OsMYB55 is involved in the tolerance to high temperature and modulates amino acid metabolism. PLoS One 7(12):e52030
Fahlgren N, Carrington JC (2010) miRNA target prediction in plants. Methods Mol Biol (Clifton, NJ) 592:51D57. doi:10.1007/978-1-60327-005-2_4
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, Carrington JC (2007) High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2(2):e219
Ferdous J, Sanchez-Ferrero JC, Langridge P, Milne L, Chowdhury J, Brien C, Tricker PJ (2016) Differential expression of microRNAs and potential targets under drought stress in barley. Plant Cell Environ. doi:10.1111/pce.12764
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. doi:10.1093/bioinformatics/bts565
Guerra D, Crosatti C, Khoshro HH, Mastrangelo AM, Mica E, Mazzucotelli E (2015) Post-transcriptional and post-translational regulations of drought and heat response in plants: a spider’s web of mechanisms. Frontiers in Plant Sci. doi:10-3389/fpls.2015.00057
Hackenberg M, Gustafson P, Langridge P, Shi BJ (2015) Differential expression of microRNAs and other small RNAs in barley between water and drought conditions. Plant Biotechnol J. doi:10.1111/pbi.12220
Hu H, Xiong L (2014) Genetic engineering and breeding of drought-resistant crops. Annu Rev Plant Biol 65:715–741. doi:10.1146/annurev-arplant-050213-040000
Kantar M, Lucas SJ, Budak H (2011) miRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 233(3):471–484
Khraiwesh B, Zhu JK, Zhu J (2012) Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys Acta 1819:137–148
Kinoshita T, Ono N, Hayashi Y, Morimoto S, Nakamura S, Soda M, Kato Y, Ohnishi M, Nakano T, Inoue S, Shimazaki K (2011) FLOWERING LOCUS T regulates stomatal opening. Curr Biol 21:1232–1238
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73. doi:10.1093/nar/gkt1181
Kumimoto RW, Adam L, Hymus GJ, Repetti PP, Reuber TL, Marion CM, Hempel FD, Ratcliffe OJ (2008) The Nuclear Factor Y subunits NF-YB2 and NF-YB3 play additive roles in the promotion of flowering by inductive long-day photoperiods in Arabidopsis. Planta 228:709–723
Kurihara Y, Watanabe Y (2004) Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci U S A 101:12753–12758
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi:10.1186/gb-2009-10-3-r25
Lee M et al (2008) The ABC transporter AtABCB14 is a malate importer and modulates stomatal response to CO2.Nat. Cell Biol 10(10):1217–1223
Levitt J (1980) Response of plants to environmental stresses. Water, Salt and other Stresses. Academic Press NY 1: 129-186.
Li WX, Oono Y, Zhu J, He XJ, Wu JM, Iida K, Lu XY, Cui X, Jin H, Zhu JK (2008) The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttransciptionally to promote drought resistance. Plant Cell 20:2238–2251
Liu H, Searle IR, Watson-Haigh NS, Baumann U, Mather DE, Able AJ, Able JA (2015) Genome-wide identification of microRNAs in leaves and the developing head of four durum genotypes during water deficit stress. PLoS One 10:e0142799. doi:10.1371/journal.pone.0142799
Liu H, Able AJ, Able JA (2016) Water-deficit stress-responsive microRNAs and their targets in four durum wheat genotypes. Funct Integr Genomics. doi: 10.1007/s10142-016-0515-y.
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Ma X, Xin Z, Wang Z, Yang O, Guo S, Guo X, Cao L, Lin T (2015) Identification and comparative analysis of differentially expressed miRNAs in leaves of two wheat genotypes during dehydration stress. BMC Plant Biol 15:21
Martin M (2011) Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet journal 17:10–12
Mcginnis S, Madden TL (2004) BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 32:W20–W25
Mekonnen DW, Flügge UI, Ludewig F (2016) Gamma-aminobutyric acid depletion affects stomata closure and drought tolerance of Arabidopsis thaliana. Plant Sci 245:25–34. doi:10.1016/j.plantsci.2016.01.005
Merlot S, Leonhardt N, Fenzi F, Valon C, Costa M, Piette L, Vavasseur A, Genty B, Boivin K, Muller A, Giraudat J, Leung J (2007) Constitutive activation of a plasma membrane H(+)-ATPase prevents abscisic acid-mediated stomatal closure. EMBO J 26(13):3216–3226
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qi Y, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu J-K (2008) Criteria for annotation of plant MicroRNAs. Plant Cell 20:3186–3190
Moshelion M, Halperin O, Wallach R, Oren R, Way D (2015) Role of aquaporins in determining transpiration and photosynthesis in water-stressed plants: crop water-use efficiency, growth and yield. Plant Cell Environ 38:1785–1793
Panio G, Motzo R, Mastrangelo AM, Marone D, Cattivelli L, Giunta F, De Vita P (2013) Molecular mapping of stomatal-conductance-related traits in durum wheat (Triticum turgidum ssp. durum). Ann Appl Biol 162:258–270
Phookaew P, Netrphan S, Sojikul P, Narangajavana J (2014) Involvement of miR164- and miR167-mediated target gene expressions in responses to water deficit in cassava. Biol Plantarum 58:469–478. doi:10.1007/s10535-014-0410-0
Ren Y, Chen L, Zhang Y, Kang X, Zhang Z, Wang Y (2012) Identification of novel and conserved Populus tomentosa microRNA as components of a response to water stress. Funct Integr Genom 12:327–339
Reyes JL, Chua NH (2007) ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J 49:592–606
Rizza F, Ghashghaie J, Meyer S, Matteu L, Mastrangelo AM, Badeck FW (2012) Constitutive differences in water use efficiency between two durum wheat cultivars. Field Crop Res 125:49–60
Rogers K, Chen X (2013) Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 25:2383–2399. doi:10.1105/tpc.113.113159
Sunkar R, Li YF, Jagadeeswaran G (2012) Functions of microRNAs in plant stress responses. Trends Plant Sci 17:196–203
Tardieu F, Simonneau T (1998) Variability among species of stomatal control under fluctuating soil water status and evaporative demand: modelling isohtdric and anisohydric behaviours. J Exp Bot 49:419–432
Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP (2007) Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3:12
Wang Y, Sun F, Cao H, Peng H, Ni Z, Sun Q, Yao Y (2012) TamiR159 directed wheat TaGAMYB cleavage and its involvement in anther development and heat response. PLoS One 7(11):e48445
Wang Y, Noguchi K, Ono N, Inoue S, Terashima I, Kinoshita T (2013) Overexpression of plasma membrane H + -ATPase in guard cells promotes light-induced stomatal opening and enhances plant growth. Proc Natl Acad Sci U S A 111:533–538
Windels D, Vazquez F (2011) miR393: integrator of environmental cues in auxin signaling? Plant Signal Behav 6(11):1672–1675
Wu G, Poethig S (2006) Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 133:3539–3547
Wu L, Liu D, Wu J, Zhang R, Qin Z, Liu D, Li A, Fu D, Zhai W, Mao L (2013) Regulation of FLOWERING LOCUS T by a microRNA in Brachypodium dystachion. Plant Cell 25:4363–4377
Wu X, Ding D, Shi C, Xue Y, Zhang Z, Tang G, Tang J (2016) microRNA-dependent gene regulatory networks in maize leaf senescence. BMC Plant Biol 16:73. doi:10.1186/s12870-016-0755-y
Xia K, Wang R, Ou X, Fang Z, Tian C, Duan J, Wang Y, Zhang M (2012) OsTIR1 and OsAFB2 downregulation via OsmiR393 overexpression leads to more tillers, early flowering and less tolerance to salt and drought in rice. PLoS One 7(1):e30039. doi:10.1371/journal.pone.0030039
Xu X, Bai H, Liu C, Chen E, Chen Q, Zhuang J, Shen B (2014) Genome-wide analysis of microRNAs and their target genes related to leaf senescence of rice. PLoS One 9:e114313. doi:10.1371/journal.pone.0114313
Xu K, Chen S, Li T, Ma X, Liang X, Ding X, Liu H, Luo L (2015) OsGRAS23, a rice GRAS transcription factor gene, is involved in drought stress response through regulating expression of stress-responsive genes. BMC Plant Biol 15:141. doi:10.1186/s12870-015-0532-3
Zhang W, Gao S, Zhou X, Xia J, Chellappan P, Zhou X, Zhang X, Jin H (2010) Multiple distinct small RNAs originate from the same microRNA precursors. Genome Biol 11:R81
Zhang X, Zou Z, Gong P, Zhang J, Ziaf K, Li H, Xiao F, Ye Z (2011) Over-expression of microRNA 169 confers enhanced drought tolerance to tomato. Biotechnol Lett 33:403–409
Zhao B, Ge L, Liang R, Li W, Ruan K, Lin H, Jin Y (2009) Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol Biol 10(29) doi:10.1186/1471-2199-10-29.
Acknowledgments
This work was supported by the Italian Ministry of Education, University and Research, project “PON01_01145-ISCOCEM.”
Author contributions
LG conducted the wet lab work and data analysis. EM interpreted the bioinformatics data and wrote the manuscript. EB designed and carried out all the computational analyses for microRNA identification and differential expression analysis. AMDL prepared the plants and RNA samples. PF contributed to bioinformatics analysis. LC designed the study, contributed to the development of the project, and edited the manuscript. CC conducted the wet lab work and wrote the manuscript. All authors have read, edited, and approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article forms part of a special issue of Functional & Integrative Genomics entitled “miRNA in model and complex organisms” (Issue Editors: Hikmet Budak and Baohong Zhang)
Lorenzo Giusti and Erica Mica contributed equally to this work.
Rights and permissions
About this article

Cite this article
Giusti, L., Mica, E., Bertolini, E. et al. microRNAs differentially modulated in response to heat and drought stress in durum wheat cultivars with contrasting water use efficiency. Funct Integr Genomics 17, 293–309 (2017). https://doi.org/10.1007/s10142-016-0527-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-016-0527-7