
Abstract
MicroRNAs, or miRNAs, are posttranscriptional regulators of gene expression. A wealth of observations and findings suggest highly complex, multicomponent, and intermingled pathways governing miRNA biogenesis and miRNA-mediated gene silencing. Plant miRNA genes are usually found as individual entities scattered around the intergenic and—to a much lesser extent—intragenic space, while miRNA gene clusters, formed by tandem or segmental duplications, also exist in plant genomes. Genome duplications are proposed to contribute to miRNA family expansions, as well. Evolutionarily young miRNAs retaining extensive homology to their loci of origin deliver important clues into miRNA origins and evolution. Additionally, imprecisely processed miRNAs evidence noncanonical routes of biogenesis, which may affect miRNA expression levels or targeting capabilities. Majority of the knowledge regarding miRNAs comes from model plant species. As ongoing research progressively expands into nonmodel systems, our understanding of miRNAs and miRNA-related pathways changes which opens up new perspectives and frontiers in miRNA research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are a class of small, endogenous, noncoding RNAs with a big impact on virtually all biological processes. MiRNAs mediate posttranscriptional gene silencing and have well-established roles in the regulation of key processes, such as development, growth, and stress responses (Budak et al. 2014, 2015b). In plants, miRNA research has been ongoing for a little over a decade; while some aspects of miRNAs, such as their biogenesis, have been unveiled in great detail, some other aspects, such as their origins, remain elusive. A thorough understanding of miRNA function enables researchers to dissect molecular mechanisms and design exogenous interventions to modulate gene expression. MiRNA research also provides clues into the evolutionary events, which may even be indicative of major episodes of phenotypic evolution, such as the emergence of plants adapted to land (Taylor et al. 2014). Here, we briefly describe canonical and noncanonical biogenesis routes of miRNAs, explore their genomic organization and miRNA gene families, and discuss their origins and evolution.
Canonical and noncanonical miRNA biogenesis
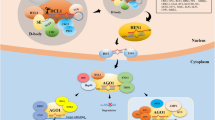
Canonical miRNA biogenesis begins with the transcription of miRNA (MIR) genes by RNA Polymerase II (Pol II, Fig. 1). The recruitment of Pol II to MIR promoters involves the interaction of several transcriptional activators and various sequence motifs in MIR promoters, indicating a multicomponent mode of regulation of MIR transcription (Rogers and Chen 2013). Primary miRNA transcripts, or pri-miRNAs, generated by transcription of the MIR loci, then fold into hairpin structures which are recognizable by the members of Dicer-like (DCL) family enzymes. The size of DCL family varies in different plant species. The DCL family has four members in Arabidopsis and grape, five members in rice and sorghum, and only three members in the moss Physcomitrella patens (Liu et al. 2009). Of the DCL family, DCL1 is mainly responsible for the cleavage of the pri-miRNA into a precursor-miRNA (pre-miRNA) in Arabidopsis. DCL1 also carries out the subsequent cleavage of pre-miRNA to release the miRNA/miRNA* duplex (Axtell et al. 2011). Different DCL family members give rise to miRNAs of different lengths. Majority of the plant miRNAs are 21 nucleotides in length, which are either processed by DCL1 or DCL4. DCL2 and DCL3 proteins, on the other hand, tend to generate miRNAs that are 22 and 24 nucleotides long, respectively. The molecular structures of DCL proteins, in particular the distance between the RNase III and PAZ domains, are suggested to be the major determinant of the resulting miRNA length (Liu et al. 2009; Rogers and Chen 2013).

An overview of canonical miRNA biogenesis, with noncanonical routes. miRNA biogenesis begins with transcription of MIR loci by RNA polymerase II. 21-nucleotide long canonical miRNAs are mostly processed by Dicer-like 1 (DCL1), while DCL2, DCL3, and DCL4 generate miRNAs of differing lengths. miRNA/miRNA* duplexes are methylated by HEN1 for stabilization, which may also contribute to the export. The export of miRNAs is generally attributed to HASTY, although this is challenged by hst mutants. In contrast to the canonical base-to-loop processing, miRNAs can also be processed from loop to base
The generation of the miRNA/miRNA* duplex occurs inside the nucleus within specialized compartments, called Dicing-bodies or D-bodies, in plants. The miRNA/miRNA* modified at the 3′ terminus by methylation by Hua Enhancer 1 (HEN1) is then exported to the cytoplasm, possibly through HASTY (Axtell et al. 2011). However, hst mutants did not exhibit accumulation of miRNAs inside the nucleus in Arabidopsis, suggesting additional export mechanisms that are yet-to-be-identified (Park et al. 2005). 2′-O-methylation of miRNA/miRNA* duplex by HEN1 is crucial to protect the 3′ termini of unwound mature miRNA from the action of exonucleases, such as small RNA-degrading nuclease (SDN) proteins (Ramachandran and Chen 2008). Otherwise, miRNA is rapidly uridylated by terminal uridyl-transferases and subsequently degraded (Yu et al. 2005; Ren et al. 2015). Methylation of the miRNA/miRNA* duplex by HEN1 may also occur in the cytoplasm as HEN1 is found both inside the nucleus and in the cytoplasm.
Once inside the cytoplasm, miRNA/miRNA* duplex separates, and the guide strand is loaded into the RNA-induced silencing complex through binding with Argonaute (AGO) proteins. The selection of the guide strand is at least partially dependent upon the thermodynamic stabilities of the 5′ ends, which is also linked to AGO binding. Most plant miRNAs carry 5′ U (uridine) that is usually bound by AGO1. The fully assembled RISC, which includes additional proteins such as Heat Shock Protein 90 (Hsp90), then binds to its target through sequence complementarity with its mature miRNA strand, to direct either mRNA cleavage or translational inhibition (Rogers and Chen 2013; Budak et al. 2015b). Although mRNA cleavage is the most prominent form of gene regulation executed by plant miRNAs, some of the major processes such as key developmental switches are controlled by translational inhibition. For instance, floral determinacy is largely regulated by the miR172 family through translational inhibition of APETALA 2 in Arabidopsis; this switch from meristem tissue to floral organs is a key to the reproductive success (Chen 2004). The regulation of APETALA2 expression by the miR172 family itself presents a robust but complex mechanism for floral development. Certain members of the miR172 are activated by POWERDRESS (PWR) protein, which do not affect yet other miR172 members, conferring this family a versatile functional redundancy to effectively control the development of floral organs (Yumul et al. 2013). These observations clearly demonstrate the intricate network of miRNA biogenesis and miRNA-regulated gene expression in plant systems (Appels et al. 2015).
In the canonical route of miRNA biogenesis, pre-miRNAs are processed from the base of the hairpin toward the loop by the DCL1. As a subtle modification of this process, miRNAs being processed in the reverse orientation, i.e., from the loop toward the base, have been reported (Fig. 1). For instance, miR319 and miR159 in Arabidopsis were observed to be processed in this reverse orientation, which requires a great degree of sequence complementarity in the primary miRNA molecule (Bologna et al. 2009). Among these loop-to-base processed pri-miRNAs, pri-miR319 was shown to generate additional small RNA species from the loop-proximal part of its stem. Even though these small RNAs were named as miRNA-like RNAs when they first described, one of the them, miR319b.2, was recognized as a miRNA after the elucidation of its miRNA* accumulation and functional properties (Budak et al. 2015a; Sobkowiak et al. 2012). Intriguingly, bidirectional processing of pri-miRNAs for miR166 family by DCL1 has been reported, leading to productive and abortive miRNA processing. The base-to-loop and loop-to-base processing was suggested to accumulate and destroy the miR166 levels, respectively, in Arabidopsis (Zhu et al. 2013).
In animals, miRNAs deriving from intronic sequences of protein-coding genes bypass Drosha cleavage, representing the Drosha-independent noncanonical biogenesis pathway. In plants, this novel class of miRNAs, called the mirtrons, also skips the first cleavage step by DCL1. Instead, splicing and subsequent debranching of the lariat generate the pre-miRNA molecule which is indentified through the hallmark of splice patterns (Axtell et al. 2011; Meng and Shao 2012).
Plant pri-miRNAs vary considerably in length, from tens to hundreds of bases, which are nonetheless processed by the miRNA biogenesis machinery. Long inverted repeat transcripts, however, appear to spawn an array of imprecisely processed miRNA species through the action of mostly noncanonical DCL proteins, DLC2, DCL3, and DCL4 (Fig. 1). These noncanonical pathways, sometimes converging to or overlapping with the siRNA-related pathways, are still poorly understood in plants; however, they may play roles in the birth of new miRNAs (Axtell et al. 2011).
Organellar miRNAs
Organellar miRNAs that reside in mitochondria and chloroplasts are an emerging issue in miRNA research, as they raise up the possibility of extant miRNA species encoded and processed by the genomes of these organelles. Recent studies identified miRNA co-localization with mitochondria in rats, mice, and humans (Kren et al. 2009; Bian et al. 2010; Barrey et al. 2011; Sripada et al. 2012a). Isolated mitochondria revealed differential enrichment of miRNAs, independent of cellular miRNAs, and the presence of AGO2 protein, which together suggest modulation of mitochondria functions through miRNAs (Bian et al. 2010; Das et al. 2012). While miRNAs were in silico identified from or mapped to the mitochondrial DNA (mtDNA), the origins of the miRNAs residing in the mitochondria, collectively termed “mitomiRs” could not be determined (Barrey et al. 2011; Bandiera et al. 2011). Although the translocation of nuclear-encoded miRNAs into the mitochondria has been shown, whether miRNAs transcribed from the mtDNA exist remains an intriguing question (Barrey et al. 2011; Das et al. 2012). Considering that Arabidopsis mutants that do not express HASTY, the protein attributed to miRNA export from the nucleus, did not accumulate miRNAs inside the nucleus (Park et al. 2005), miRNA translocation into the double-membrane-enclosed mitochondria represents another intriguing aspect of mitomiRs. A candidate for the miRNA import system in mitochondria has been suggested as an intermembrane space protein, polynucleotide phosphorylase (PNPASE). This protein is conserved across species from bacteria to humans and is also found in plant mitochondria and chloroplasts (Sripada et al. 2012b). Clues from mitomiRs can reveal noncanonical biogenesis routes or offer additional players for miRNA export from nucleus.
The presence of small, noncoding RNAs inside the chloroplasts was first described in Arabidopsis in 2002 (Marker et al. 2002) and later reported from tobacco and cabbage (Lung et al. 2006; Wang et al. 2011). While at least a fraction of these small RNAs are likely degradation products, Wang and his colleagues demonstrated that chloroplast small RNA population was affected by heat treatment, pointing out to active mechanisms of small RNA metabolism inside this organelle. Strikingly, two heat-responsive chloroplast small RNAs, which did not originate from rRNA or tRNA species, revealed complementary sequences with two transcripts. The expression levels of these transcripts contrasted with that of the corresponding small RNAs under heat stress, suggesting miRNA-like regulation of gene expression inside chloroplasts (Wang et al. 2011).
MitomiRs and cipomiRs, named in reference to mitomiRs, are still poorly understood in plants (Budak et al. 2015b). Few reports hinder firm conclusions on whether these organelles are capable of organelle-specific miRNA biogenesis or they have specialized systems for miRNA import. In each case, further research is likely to contribute to the elucidation of noncanonical routes for miRNA processing and transport.
Identification of miRNAs
Earliest attempts for miRNA identification included cloning and sequencing of small RNAs (sRNAs) (Llave et al. 2002; Sunkar and Zhu 2004). However, as these approaches had been both time-consuming and labor-intensive, computational identification methods that utilize distinctive features of miRNAs and extensive conservation among plant miRNAs soon emerged (Wang et al. 2004; Bonnet et al. 2004). The introduction of next-generation sequencing (NGS) technologies and ever-decreasing costs of these platforms enabled genome or transcriptome-wide in silico identification of many miRNA precursors in both model and nonmodel plants (Jia et al. 2013; Ling et al. 2013; Kurtoglu et al. 2014; Budak and Kantar 2015). Additionally, chromosome-based genomics studies revealed chromosome-specific miRNA catalogues, which even allowed comparisons between the miRNA contents of homologous chromosomes in polyploid species, such as the hexaploid bread wheat (Kantar et al. 2012; Kurtoglu et al. 2013; Deng et al. 2014). While computational techniques are powerful approaches for high-throughput miRNA identification, in silico predicted miRNAs require additional experimental evidence for verification. Quantitative real-time-polymerase chain reaction (qRT-PCR), Northern blotting, and microarrays are among the most commonly used techniques to provide experimental evidence of expression for computationally predicted miRNAs (Wang et al. 2004; Ren et al. 2012; Kurtoglu et al. 2013; Budak and Akpinar 2011). However, they are not very sensitive in terms of identifying the exact mature miRNA sequence, failing to differentiate isomiRs and the miRNAs belonging to the same miRNA family. Another application of NGS technologies for high-throughput miRNA identification is deep-sequencing of sRNA libraries. Even though this technique is highly powerful in terms of sequence specificity, appropriate precursor sequences should be provided for the verification of genuine miRNAs, which is mostly performed through in silico analyses.
Plant miRNAs require strict sequence complementarity with their target transcripts, only allowing a few mismatches (Zhao et al. 2015). Therefore, in addition to miRNAs and their precursors, miRNA targets can be confidently identified through computational methods in plants. Although a direct link between a miRNA and its target should be established through experimental procedures such as rapid amplification of cDNA ends (RACE) or degradome sequencing, identification and annotation of putative miRNA targets are broadly indicative of the physiological roles of the corresponding miRNAs (Budak et al. 2015b). An overview of high-throughput miRNA identification and downstream applications is depicted in Fig. 2.

A generalized scheme of high-throughput miRNA identification and subsequent analyses. Precursors and corresponding miRNAs can be identified from either genome or transcriptome-wide or chromosome-specific sequence data that may or may not be masked against repeat elements, as a fraction of miRNAs are encoded by the repetitive regions. The identification of isomiRs, sequence variants of mature miRNAs, and miRNA targets are now common downstream applications to elucidate the structures and functions of miRNAs of interest. EST expressed sequence tag, RACE rapid amplification of cDNA ends, RNA-Seq RNA-sequencing
Genomic organization of miRNA genes
Plant miRNAs have been suggested to form large miRNA gene families, in contrast to the animal miRNAs. This notion changes as more advanced techniques such as deep sequencing of sRNA libraries enable high-throughput identification of large numbers of miRNAs. According to the current version of the miRBase miRNA registry (http://www.mirbase.org/, Release 21), miRNA gene family sizes are smaller than those reported previously (Li and Mao 2007), although part of this reduction can be explained by spurious miRNA entries which lack proper verification. The average size of the miRNA gene families, as deposited in miRBase, is similar across different plant species, except for maize, Zea mays, grape, Vitis vinifera, and sorghum, Sorghum bicolor, with average miRNA family sizes of 5.93, 3.40, and 3.25, respectively (Table 1). The miRNA families associated with development and/or stress responses, such as miR156, miR169, and miR395, are generally represented by large families with several members in different plants (Chuck et al. 2010; Liang et al. 2012; Wang et al. 2013; Sorin et al. 2014). Interestingly, the miR2592 family with 66 members in Medicago truncatula (Table 1) is deemed as legume-specific and has been reported from a few species so far, with roles in organogenesis and stress response (Zhou et al. 2012; Liu et al. 2014). However, recent studies have shown that miR2592 responds to heat stress in radish (Wang et al. 2014). Despite these large miRNA families with established roles in various pathways, the conservation status of miRNA families, i.e., conserved and nonconserved families, along with average miRNA family sizes across species may change depending on new findings.
Plant miRNAs are mostly transcribed from independent, non-protein-coding loci on the intergenic regions of the genome. Plant miRNA genes can also be found in clusters; however, these clusters mostly code for homologous miRNA species, in contrast to animals where multiple miRNA species can be transcribed as a single polycistronic transcript from a cluster (Li and Mao 2007). This pattern suggests that most plant miRNA gene clusters are formed through tandem duplications linked to dosage effect, such that co-transcription from the cluster would increase the dosage of a specific miRNA at once (Axtell et al. 2011). Additionally, segmental duplications are also indicted in the formation of homologous miRNA gene clusters in plants, such as miR395 family, which is known to form clusters in several plant species (Li and Mao 2007; Sun et al. 2012). Few gene clusters coding for nonhomologous miRNAs have also been reported (Merchan et al. 2009; Cui et al. 2009). Nonhomologous miRNAs within a single cluster were observed to target different proteins belonging exclusively to the same protein family. Thus, while homologous miRNA gene clusters are associated with dosage effect, nonhomologous miRNA gene clusters are implicated in the co-regulation of related proteins (Merchan et al. 2009).
Besides the intergenic miRNA loci, intronic miRNAs or the so-called “mirtrons,” are being increasingly reported in plants. Mirtrons were first discovered in animals through the splicing-patterned ends of pre-miRNAs, in contrast to the canonical Drosha-processed ends (Ruby et al. 2007). So far, mirtrons, transcribed from the spliced out introns of the protein-coding genes, have been reported in the model plants Arabidopsis thaliana and rice, as well as nonmodel plants such as cassava and foxtail millet (Meng and Shao 2012; Joshi et al. 2012; Yi et al. 2013; Patanun et al. 2013). Interestingly, an intronic miRNA, miR838, had been located on intron 14 of the DCL1 primary transcript in Arabidopsis, suggesting a feedback loop for the autoregulation of miRNA biogenesis (Rajagopalan et al. 2006). Despite being rare, exonic miRNAs, transcribed from the exons of protein-coding genes, have also been described in plants (Li et al. 2011a; Colaiacovo et al. 2012; Liu 2012). These exonic miRNAs are suspected to take part in fine-tuning of the expression of their targets (Li et al. 2011a).
Origins of plant miRNAs
The requirement for near-perfect complementarity for target recognition facilitates in silico identification of putative targets of miRNAs in plants. This requirement, as well as close evolutionary dynamics of miRNAs and their targets (Zhao et al. 2015), suggests that the origins of plant miRNAs are linked to their cognate targets. An inverted duplication of a gene copy is capable of producing a hairpin structure that can be processed to generate miRNAs (Cuperus et al. 2011). It is speculated that at the initial stages, these hairpins are imprecisely processed by a number of DCL enzymes, particularly DCL2, DCL3, and DCL4 besides the canonical DCL1, to generate small RNAs of varying sizes. This way, the most convenient miRNA-target pair is selected along the evolution without the action of an immediate selection pressure (Axtell et al. 2011). Accordingly, recently evolved plant miRNA genes exhibiting extensive homology to their targets have been reported, indicating that these miRNAs might have evolved from the duplicated copies of their targets (Nozawa et al. 2012; He et al. 2014). In fact, miR826 and miR5090 have been suggested to derive from the same transcript in Arabidopsis (He et al. 2014). Additionally, rice miR7695 is likely evolved from a duplication of its target gene, due to the high similarity to its target and DCL4—instead of the canonical DCL1—processing during its biogenesis (Campo et al. 2013). Once a miRNA gene is evolved, tandem or segmental duplications are responsible for the expansion of miRNA gene families.
Transposable elements (TEs) are another major source of miRNA formation in plants. TEs further contribute to miRNA gene family expansions through tandem and segmental duplications. Plant miRNAs originating from TEs, also called TE-MIRs, have been well-documented, although converging miRNA and siRNA pathways obscure the identification of bona fide miRNAs among them (Li et al. 2011b; Kurtoglu et al. 2014). Interestingly, TE-MIRs may be promoted in some plant species, where they make up the majority of the overall miRNA content. Sun and her colleagues observed that, among four well-studied species, TE-derived miRNAs were highly abundant in Populus trichocarpa and Oryza sativa compared to Sorghum bicolor and Arabidopsis; whether this apparent bias in TE-MIRs has evolutionary implications remains unclear at this time (Sun et al. 2012). In the formation of young miRNAs, the nonautonomous miniature inverted-repeat transposable elements (MITEs) are of particular importance due to their palindromic structures (Cuperus et al. 2011; Sun et al. 2012). A MITE element inserted within the promoter region of an important vernalization gene, VrnA1a, contained the sequence of TamiR1123 in wheat, whose expression correlated with the vernalization gene (Yu et al. 2014). TE-MIRs were observed to frequently associate with intragenic regions in monocots among four test species. As MITEs preferentially insert into genic regions, monocots may contain a higher proportion of MITE-related miRNAs compared to eudicots (Sun et al. 2012).
Occasionally, plant miRNAs arise from random unstructured sequences (de Felippes et al. 2008). While this route is common for new miRNAs to emerge in animals where they can “creep” into the regulatory system, plant miRNAs arising from random hairpins are prone to be lost quickly due to the lack of fortuitous targets. Due to high-sequence complementarity necessary to target a gene, plant miRNAs from random sequences rarely acquire targets that would stably integrate them into the miRNA regulatory network (Axtell et al. 2011). In Arabidopsis, miR447 and miR856 had validated targets that were distinct from the fold back sequences they were processed from, representing the rare subclass of miRNAs originating from random unstructured sequences (Fahlgren et al. 2007).
The spliced-out introns of protein-coding genes are increasingly recognized to spawn miRNAs, the so-called mirtrons, in plants, as they do in animals. Interestingly, mirtrons identified in Arabidopsis and rice revealed a bias for 24-nt long species, beginning with 5′-G (guanine) and 5′-A (adenosine), in contrast to the 21-nt long canonical miRNAs that generally begins with 5′-U (uridine) (Meng and Shao 2012). These features point out to noncanonical processing of mitrons. Curiously, some of the mirtrons identified in this study were found to reside in TE genes (Meng and Shao 2012). Mirtrons are an emerging issue in plants; further studies should contribute to our understanding of miRNA origins and evolution.
The origins of miRNAs are mostly traced back through young miRNAs that, unlike the ancient miRNAs conserved across many species, retain considerable similarity to their loci of origin. As these young miRNAs are likely evolved relatively recently, most of them are species-specific and are suggested to be gained and lost at high frequencies. Additionally, these young miRNAs often have low expression levels and exhibit improper processing, which may indicate a transition state before complete integration into the regulatory system (Cuperus et al. 2011). However, besides a few model species, miRNA research is still in infancy for most species, and serious concerns are increasingly raised for spurious miRNAs deposited in public databases. As another intriguing aspect of young miRNAs, the integration of new miRNAs into the canonical miRNA network appears to happen through miRNAome expansions at distinct evolutionary episodes, which in theory may coincide with phenotypic evolution in plants (Taylor et al. 2014). Thus, our views on the “young” miRNAs may change, thereby changing our view on miRNA evolution.
Evolution of plant miRNAs
The emergence of gene silencing through complementary RNA molecules is considered as an evolutionarily ancient event, which was lost in certain eukaryotic lineages, such as yeast, along the way. In fact, two miRNAs, miR854 and miR855, are common to both animal and plant kingdoms, although there is controversy on the authenticity of these miRNAs (Arteaga-Vázquez et al. 2006; Cuperus et al. 2011; Pashkovskiy and Ryazansky 2013). However, evolutionary conclusions regarding conserved and nonconserved miRNA families should be drawn cautiously, because other than a few model species, miRNAs are not extensively characterized in many plants and several of which reported so far still require additional evidence for verification. To demonstrate this problem, Taylor and his colleagues analyzed a total of 6172 miRNA genes deposited in v20 of miRBase in detail, and almost one third of these miRNAs lacked enough evidence to be annotated as genuine miRNAs. Additionally, as the same study points out, there are large evolutionary gaps between species for which miRNA content information is available. For instance, Physcomitrella patens is the only representative in miRBase from the moss lineage (Taylor et al. 2014). Consequently, our understanding of the evolutionarily conserved and nonconserved miRNA families across evolutionary lineages is prone to change as the miRNA catalogues of more species are unlocked.
Nevertheless, eight miRNA families, namely, miR156, miR159/319, miR160, miR166, miR171, miR408, miR390/391, and miR395, were found to be conserved across the ancestral Embryophyta (Cuperus et al. 2011). Of these, miR156 and miR166 are concluded as conserved miRNAs across the plant kingdom that have roles in flower development (Luo et al. 2013). Additionally, miR396 family was found in all vascular plants, and miR397 and miR398 families were present in all seed plants. Among the angiosperms, miR403, miR828, and miR2111 families are so far known as eudicot-specific (Cuperus et al. 2011). Interestingly, targets of miR403 family include AGO2 and AGO3 proteins, indicating a feedback look in miRNA biogenesis (Jagtap and Shivaprasad 2014).
While the lists of conserved and nonconserved miRNA families may change depending on new findings, syntenic relationships may corroborate that miRNAs conserved across two or more species are in fact representative of a true evolutionary conservation event. For instance, miR1133 and miR167 identified from the short arm of chromosome 5D of bread wheat, Triticum aestivum, have their homologs on the syntenic Brachypodium distachyon chromosome 4, strongly indicating that these miRNAs are likely conserved between these species (Kurtoglu et al. 2013). In conclusion, evolutionary patterns of miRNA conservation can be misleading at times, unless the miRNAs themselves or the relationship between the species is supported by additional lines of evidence.
References
Appels R, Nystrom J, Webster H, Keeble-Gagnere G (2015) Discoveries and advances in plant and animal genomics. Funct Integr Genomics. doi:10.1007/s10142-015-0434-3
Arteaga-Vázquez M, Caballero-Pérez J, Vielle-Calzada J-P (2006) A family of microRNAs present in plants and animals. Plant Cell 18:3355–3369. doi:10.1105/tpc.106.044420
Axtell MJ, Westholm JO, Lai EC (2011) Vive la différence: biogenesis and evolution of microRNAs in plants and animals. Genome Biol 12:221. doi:10.1186/gb-2011-12-4-221
Bandiera S, Rüberg S, Girard M et al (2011) Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One 6:e20746. doi:10.1371/journal.pone.0020746
Barrey E, Saint-Auret G, Bonnamy B et al (2011) Pre-microRNA and mature microRNA in human mitochondria. PLoS One 6:e20220. doi:10.1371/journal.pone.0020220
Bian Z, Li L-M, Tang R et al (2010) Identification of mouse liver mitochondria-associated miRNAs and their potential biological functions. Cell Res 20:1076–1078. doi:10.1038/cr.2010.119
Bologna NG, Mateos JL, Bresso EG, Palatnik JF (2009) A loop-to-base processing mechanism underlies the biogenesis of plant microRNAs miR319 and miR159. EMBO J 28:3646–3656. doi:10.1038/emboj.2009.292
Bonnet E, Wuyts J, Rouzé P, Van de Peer Y (2004) Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proc Natl Acad Sci U S A 101:11511–11516. doi:10.1073/pnas.0404025101
Budak H, Akpinar BA (2011) Dehydration stress-responsive miRNA in Brachypodium distachyon: evident by genome-wide screening of microRNAs expression. OMICS J Integr Biol 15(11):791–799. doi:10.1089/omi.2011.0073
Budak H, Kantar M (2015) Harnessing NGS and big data optimally: comparison of miRNA prediction from assembled versus non-assembled sequencing data—the case of the grass Aegilops tauschii complex genome. OMICS: International Journal of Plant Biology
Budak H, Khan Z, Kantar M (2014) History and current status of wheat miRNAs using next-generation sequencing and their roles in development and stress. Brief Funct Genomics. doi:10.1093/bfgp/elu021
Budak H, Bulut R, Kantar M, Alptekin B (2015a) MicroRNA nomenclature and the need for a revised naming prescription. Brief Funct Genomics
Budak H, Kantar M, Bulut R, Akpinar BA (2015b) Stress responsive miRNAs and isomiRs in cereals. Plant Sci in press:1–13. doi:10.1016/j.plantsci.2015.02.008
Campo S, Peris-Peris C, Siré C et al (2013) Identification of a novel microRNA (miRNA) from rice that targets an alternatively spliced transcript of the Nramp6 (Natural resistance-associated macrophage protein 6) gene involved in pathogen resistance. New Phytol 199:212–227. doi:10.1111/nph.12292
Chen X (2004) A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303:2022–2025. doi:10.1126/science.1088060
Chuck G, Whipple C, Jackson D, Hake S (2010) The maize SBP-box transcription factor encoded by tasselsheath4 regulates bract development and the establishment of meristem boundaries. Development 137:1243–1250. doi:10.1242/dev.048348
Colaiacovo M, Lamontanara A, Bernardo L et al (2012) On the complexity of miRNA-mediated regulation in plants: novel insights into the genomic organization of plant miRNAs. Biol Direct 7:15. doi:10.1186/1745-6150-7-15
Cui X, Xu SM, Mu DS, Yang ZM (2009) Genomic analysis of rice microRNA promoters and clusters. Gene 431:61–66. doi:10.1016/j.gene.2008.11.016
Cuperus JT, Fahlgren N, Carrington JC (2011) Evolution and functional diversification of MIRNA genes. Plant Cell 23:431–442. doi:10.1105/tpc.110.082784
Das S, Ferlito M, Kent OA et al (2012) Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res 110:1596–1603. doi:10.1161/CIRCRESAHA.112.267732
de Felippes FF, Schneeberger K, Dezulian T et al (2008) Evolution of Arabidopsis thaliana microRNAs from random sequences. RNA 14:2455–2459. doi:10.1261/rna.1149408
Deng P, Nie X, Wang L et al (2014) Computational identification and comparative analysis of miRNAs in wheat group 7 chromosomes. Plant Mol Biol Report 32:487–500. doi:10.1007/s11105-013-0669-x
Fahlgren N, Howell MD, Kasschau KD et al (2007) High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2:e219. doi:10.1371/journal.pone.0000219
He H, Liang G, Li Y et al (2014) Two young MicroRNAs originating from target duplication mediate nitrogen starvation adaptation via regulation of glucosinolate synthesis in Arabidopsis thaliana. Plant Physiol 164:853–865. doi:10.1104/pp. 113.228635
Jagtap S, Shivaprasad PV (2014) Diversity, expression and mRNA targeting abilities of Argonaute-targeting miRNAs among selected vascular plants. BMC Genomics 15:1049. doi:10.1186/1471-2164-15-1049
Jia J, Zhao S, Kong X et al (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496:91–95. doi:10.1038/nature12028
Joshi PK, Gupta D, Nandal UK et al (2012) Identification of mirtrons in rice using MirtronPred: a tool for predicting plant mirtrons. Genomics 99:370–375. doi:10.1016/j.ygeno.2012.04.002
Kantar M, Akpınar BA, Valárik M et al (2012) Subgenomic analysis of microRNAs in polyploid wheat. Funct Integr Genomics 12:465–479. doi:10.1007/s10142-012-0285-0
Kren BT, Wong PY-P, Sarver A et al (2009) MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol 6:65–72. doi:10.4161/rna.6.1.7534
Kurtoglu KY, Kantar M, Lucas SJ, Budak H (2013) Unique and conserved microRNAs in wheat chromosome 5D revealed by next-generation sequencing. PLoS One 8:e69801. doi:10.1371/journal.pone.0069801
Kurtoglu KY, Kantar M, Budak H (2014) New wheat microRNA using whole-genome sequence. Funct Integr Genomics. doi:10.1007/s10142-013-0357-9
Li A, Mao L (2007) Evolution of plant microRNA gene families. Cell Res 17:212–218. doi:10.1038/sj.cr.7310113
Li T, Li H, Zhang Y-X, Liu J-Y (2011a) Identification and analysis of seven H2O2-responsive miRNAs and 32 new miRNAs in the seedlings of rice (Oryza sativa L. ssp. indica). Nucleic Acids Res 39:2821–2833. doi:10.1093/nar/gkq1047
Li Y, Li C, Xia J, Jin Y (2011b) Domestication of transposable elements into MicroRNA genes in plants. PLoS One 6:e19212. doi:10.1371/journal.pone.0019212
Liang G, He H, Yu D (2012) Identification of nitrogen starvation-responsive microRNAs in Arabidopsis thaliana. PLoS One 7:e48951. doi:10.1371/journal.pone.0048951
Ling H-Q, Zhao S, Liu D et al (2013) Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496:87–90. doi:10.1038/nature11997
Liu Q (2012) Novel miRNAs in the control of arsenite levels in rice. Funct Integr Genomics 12:649–658. doi:10.1007/s10142-012-0282-3
Liu Q, Feng Y, Zhu Z (2009) Dicer-like (DCL) proteins in plants. Funct Integr Genomics 9:277–286. doi:10.1007/s10142-009-0111-5
Liu W, Yu W, Hou L et al (2014) Analysis of miRNAs and their targets during adventitious shoot organogenesis of Acacia crassicarpa. PLoS One 9:e93438. doi:10.1371/journal.pone.0093438
Llave C, Kasschau KD, Rector MA, Carrington JC (2002) Endogenous and silencing-associated small RNAs in plants. 14:1605–1619. doi: 10.1105/tpc.003210.ruses
Lung B, Zemann A, Madej MJ et al (2006) Identification of small non-coding RNAs from mitochondria and chloroplasts. Nucleic Acids Res 34:3842–3852. doi:10.1093/nar/gkl448
Luo Y, Guo Z, Li L (2013) Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev Biol 380:133–144. doi:10.1016/j.ydbio.2013.05.009
Marker C, Zemann A, Terhörst T et al (2002) Experimental RNomics. Curr Biol 12:2002–2013. doi:10.1016/S0960-9822(02)01304-0
Meng Y, Shao C (2012) Large-scale identification of mirtrons in Arabidopsis and rice. PLoS One 7:e31163. doi:10.1371/journal.pone.0031163
Merchan F, Boualem A, Crespi M, Frugier F (2009) Plant polycistronic precursors containing non-homologous microRNAs target transcripts encoding functionally related proteins. Genome Biol 10:R136. doi:10.1186/gb-2009-10-12-r136
Nozawa M, Miura S, Nei M (2012) Origins and evolution of microRNA genes in plant species. Genome Biol Evol 4:230–239. doi:10.1093/gbe/evs002
Park MY, Wu G, Gonzalez-Sulser A et al (2005) Nuclear processing and export of microRNAs in Arabidopsis. Proc Natl Acad Sci U S A 102:3691–3696. doi:10.1073/pnas.0405570102
Pashkovskiy PP, Ryazansky SS (2013) Biogenesis, evolution, and functions of plant microRNAs. Biochem Biokhimii͡a 78:627–637. doi:10.1134/S0006297913060084
Patanun O, Lertpanyasampatha M, Sojikul P et al (2013) Computational identification of microRNAs and their targets in cassava (Manihot esculenta Crantz.). Mol Biotechnol 53:257–269. doi:10.1007/s12033-012-9521-z
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 20:3407–3425. doi:10.1101/gad.1476406
Ramachandran V, Chen X (2008) Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science 321:1490–1492. doi:10.1126/science.1163728
Ren Y, Chen L, Zhang Y et al (2012) Identification of novel and conserved Populus tomentosa microRNA as components of a response to water stress. Funct Integr Genomics 12:327–339. doi:10.1007/s10142-012-0271-6
Ren G, Chen X, Yu B (2015) Small RNAs meet their targets: when methylation defends miRNAs from uridylation. RNA Biol 11:1099–1104. doi:10.4161/rna.36243
Rogers K, Chen X (2013) Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 25:2383–2399. doi:10.1105/tpc.113.113159
Ruby JG, Jan CH, Bartel DP (2007) Intronic microRNA precursors that bypass Drosha processing. Nature 448:83–86. doi:10.1038/nature05983
Sobkowiak L, Karlowski W, Jarmolowski A, Szweykowska-Kulinska Z (2012) Non-canonical processing of Arabidopsis pri-miR319a/b/c generates additional microRNAs to target one RAP2.12 mRNA isoform. Front Plant Sci 3:46. doi:10.3389/fpls.2012.00046
Sorin C, Declerck M, Christ A et al (2014) A miR169 isoform regulates specific NF-YA targets and root architecture in Arabidopsis. New Phytol 202:1197–1211. doi:10.1111/nph.12735
Sripada L, Tomar D, Prajapati P et al (2012a) Systematic analysis of small RNAs associated with human mitochondria by deep sequencing: detailed analysis of mitochondrial associated miRNA. PLoS One 7:e44873. doi:10.1371/journal.pone.0044873
Sripada L, Tomar D, Singh R (2012b) Mitochondria: one of the destinations of miRNAs. Mitochondrion 12:593–599. doi:10.1016/j.mito.2012.10.009
Sun J, Zhou M, Mao Z, Li C (2012) Characterization and evolution of microRNA genes derived from repetitive elements and duplication events in plants. PLoS One 7:e34092. doi:10.1371/journal.pone.0034092
Sunkar R, Zhu J-K (2004) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16:2001–2019. doi:10.1105/tpc.104.022830
Taylor RS, Tarver JE, Hiscock SJ, Donoghue PCJ (2014) Evolutionary history of plant microRNAs. Trends Plant Sci 19:175–182. doi:10.1016/j.tplants.2013.11.008
Wang X-J, Reyes JL, Chua N-H, Gaasterland T (2004) Prediction and identification of Arabidopsis thaliana microRNAs and their mRNA targets. Genome Biol 5:R65. doi:10.1186/gb-2004-5-9-r65
Wang L, Yu X, Wang H et al (2011) A novel class of heat-responsive small RNAs derived from the chloroplast genome of Chinese cabbage (Brassica rapa). BMC Genomics 12:289. doi:10.1186/1471-2164-12-289
Wang M, Wang Q, Zhang B (2013) Response of miRNAs and their targets to salt and drought stresses in cotton (Gossypium hirsutum L.). Gene 530:26–32. doi:10.1016/j.gene.2013.08.009
Wang R, Xu L, Zhu X et al (2014) Transcriptome-wide characterization of novel and heat-stress-responsive microRNAs in radish (Raphanus sativus L.) using next-generation sequencing. Plant Mol Biol Report. doi:10.1007/s11105-014-0786-1
Yi F, Xie S, Liu Y et al (2013) Genome-wide characterization of microRNA in foxtail millet (Setaria italica). BMC Plant Biol 13:212. doi:10.1186/1471-2229-13-212
Yu B, Yang Z, Li J et al (2005) Methylation as a crucial step in plant microRNA biogenesis. Science 307:932–935. doi:10.1126/science.1107130
Yu M, Carver BF, Yan L (2014) TamiR1123 originated from a family of miniature inverted-repeat transposable elements (MITE) including one inserted in the Vrn-A1a promoter in wheat. Plant Sci 215–216:117–123. doi:10.1016/j.plantsci.2013.11.007
Yumul RE, Kim YJ, Liu X et al (2013) POWERDRESS and diversified expression of the MIR172 gene family bolster the floral stem cell network. PLoS Genet 9:e1003218. doi:10.1371/journal.pgen.1003218
Zhao M, Meyers BC, Cai C, et al (2015) Evolutionary patterns and coevolutionary consequences of MIRNA genes and MicroRNA targets triggered by multiple mechanisms of genomic duplications in soybean. Plant Cell Online 27:tpc.15.00048. doi: 10.1105/tpc.15.00048
Zhou ZS, Zeng HQ, Liu ZP, Yang ZM (2012) Genome-wide identification of Medicago truncatula microRNAs and their targets reveals their differential regulation by heavy metal. Plant Cell Environ 35:86–99. doi:10.1111/j.1365-3040.2011.02418.x
Zhu H, Zhou Y, Castillo-González C et al (2013) Bidirectional processing of pri-miRNAs with branched terminal loops by Arabidopsis Dicer-like1. Nat Struct Mol Biol 20:1106–1115. doi:10.1038/nsmb.2646
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Budak, H., Akpinar, B.A. Plant miRNAs: biogenesis, organization and origins. Funct Integr Genomics 15, 523–531 (2015). https://doi.org/10.1007/s10142-015-0451-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-015-0451-2