
Abstract
As an important economic shellfish, the pearl oyster, Pinctada fucata, and its larvae are an ideal model for studying molecular mechanisms of larval development in invertebrates. Larval development directly affects the quantity and quality of pearl oysters. MicroRNAs (miRNAs) may play important roles in development, but the effects of miRNA expression on P. fucata early development remain unknown. In this study, miRNA and mRNA transcriptomics of seven different P. fucata developmental stages were analyzed using Illumina RNA sequencing. A total of 329 miRNAs, including 87 known miRNAs and 242 novel miRNAs, and 33,550 unigenes, including 26,333 known genes and 7217 predicted new genes, were identified in these stages. A cluster analysis showed that the difference in the numbers of miRNAs was greatest between fertilized eggs and trochophores. In addition, the integrated mRNA transcriptome was used to predict target genes for differentially expressed miRNAs between adjacent developmental stages, and the target genes were subjected to a gene ontology enrichment analysis. Using the gene ontology annotation, 100 different expressed genes and 95 differentially expressed miRNAs were identified as part of larval development regulation. Real-time PCR was used to identify eight mRNAs and three miRNAs related to larval development. The present findings will be helpful for further clarifying the regulatory mechanisms of miRNA in invertebrate larval development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Early larval development greatly affects individual survival rate (Teh et al. 2012), population size (Vogeler et al. 2019), spatial distribution, and resource use during the life cycle of shellfish (Felisa et al. 2019; Niu et al. 2020). The bivalve larval developmental stages from the pelagic to benthonic phase include eggs, fertilized eggs, trochophores, D-shaped larvae, umbonal larvae, pediveliger larvae, and metamorphosed larvae (Fujimura et al. 1995; Huan et al. 2012; Xu et al. 2014). The morphological, physiological, structural, and functional characteristics of each stage are distinct and are coordinated both spatially and temporally by well-orchestrated cellular events (Kocamis et al. 2013). Microscopic studies have extensively illustrated the differences in the development of bivalve larvae (Baker and Mann 1994; Deng et al. 2020; Li et al. 2020a, b). With the recent emergence of high-throughput sequencing, the amount of bivalve-related genomic data has increased rapidly (Li et al. 2016; Song et al. 2016). Transcriptomic analytical methods have been used to study bivalve larval development, focusing on growth and development (Li et al. 2016), shell formation mechanisms (Weiss et al. 2010; Zhang et al. 2012; Liu et al. 2017), mineralization mechanisms (Addadi et al. 2010; Li et al. 2017), the digestive system (Jackson et al. 2005, 2007), the nervous system (Hadfield et al. 2000; Leise et al. 2001), and reproduction (Suquet et al. 2014; Chen et al. 2016a). However, the potential molecular mechanisms of these complex cellular processes in bivalve larval development have not been revealed owing to the currently insufficient genomic information and functional analyses.
MicroRNAs (miRNAs) are a class of highly conserved, noncoding single-stranded RNA molecules that are approximately 18–24 nucleotides in length and encoded by endogenous genes. They regulate post-transcriptional gene expression (Ambros 2004; Bartel 2004) by specifically binding the 3′-untranslated regions of mRNAs to degrade mRNAs of target genes or inhibit their translation, and they are intrinsic to early development, cell proliferation, differentiation, apoptosis, and other metabolic processes of eukaryotes (Krol et al. 2010; Zhe et al. 2016; Lopes et al. 2018; Wei et al. 2019; Yu et al. 2020; Xue et al. 2021). The miRNA and its target genes are influenced by a complex regulatory network. Multiple genes may be the targets of a single miRNA, and a single gene may be the target of multiple miRNAs (Bartel 2009). The expression levels of multiple target genes may be regulated by one miRNA or a combination of several miRNAs could fine-regulate the expression of a target gene (Malumbres 2012). Although miRNA expression levels have been investigated during the embryonic development of some marine animals (Wienholds et al. 2005; Niu et al. 2020; Wang et al. 2021a), studies of miRNA-related molecular mechanisms and embryonic developmental regulation in the pearl oyster, Pinctada fucata, are still in their infancy.
Pinctada fucata is an important economic shellfish that produces marine pearls, and it offers an ideal model for studying larval development and related molecular mechanisms in invertebrates (Li et al. 2016). Although some miRNAs involved in the regulation of pearl oyster mineralization have been reported, systemic miRNA information related to embryonic development has been relatively limited (Jiao et al. 2014; Yu et al. 2015; Zheng et al. 2016; Huang et al. 2019). In this study, miRNA and mRNA libraries from seven different larval developmental stages, egg, fertilized egg (Fer), trochophore (Tro), D-shaped (D-sha), umbonal larva (Umb), pediveliger larva (Ped), and metamorphosed larva (Met) were constructed and sequenced using Illumina Hiseq to identify miRNA expression profiles and their complementing mRNAs.
Materials and Methods
Larval Culture and Sample Collection
Larvae to be sampled at different developmental stages were first bred through artificial insemination in Beihai, Guangxi Province, China, as described by Fujimura et al. (1995). Their parents were 30 females and 30 males from a single population. A pool of sand-filtered seawater was used to house 250 million zygotes, maintained at 26.0–28.0 °C, with a salinity of 31.0–33.0‰, and a volume of approximately 20 m3. For mRNA and small RNA sequencing (RNA-seq), egg, Fer, Tro, D-sha, Umb, Ped, and Met samples were collected at 0 and 12–16 h, and 1, 10–12, 28–30, and 45–50 days after fertilization, respectively (Fig. 1). After sample collection, they were immediately cleaned with phosphate buffered saline (Thermo Fisher Scientific Inc., Pittsburgh, PA, USA) and then placed into an RNA protective solution (TaKaRa Bio Inc., Shiga, Japan) until RNA extraction.

Seven different developmental stages in P. fucata. Egg, Fer, Tro, D-sha, Umb, Ped, and Met (A–G, respectively)
RNA Extraction and cDNA Library Preparation
Total RNA was extracted using TRIzol® (Invitrogen Corp., Carlsbad, CA, USA), following the manufacturer’s instructions. Genomic DNA was removed using DNase I (TaKaRa Bio Inc.). Total RNA quality was checked using a 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA) and quantified using an ND-2000 system (NanoDrop Technologies, Inc., Wilmington, DE, USA). Then, mRNAs were purified using the PolyATtract® mRNA isolation system (Promega Corp., Madison, WI, USA). The RNeasy® RNA cleaning kit (Qiagen GmbH, Hilden, Germany) was used to concentrate samples in accordance with the manufacturer’s instructions. The syntheses of 21 paired-end libraries (seven different stages with three replicates per stage) were performed in accordance with the “TruSeq RNA Sample Preparation Guide.” Four lanes on an Illumina HiSeq2500 high-throughput sequencer (Illumina Inc., San Diego, CA, USA) were then used to sequence the prepared libraries, following the manufacturer’s instructions. The miRNA library was constructed as follows: First, 18–30 nt small RNAs were separated and recovered by PAGE electrophoresis. Then, 3′ and 5′ linker ligation was performed by formulating the reaction system. The cDNA was amplified by high-sensitivity polymerase to enrich the cDNA with both 3′ and 5′ adapters attached to expand the library yield. Finally, the target band after PCR was recovered by electrophoresis.
miRNA- and mRNA-seq
BGISEQ-500 deep-sequencing technology (BGI Genomics Co., Ltd., Shenzhen, China) was applied to sequence short reads. The removal of selected reads, which contained poly-N with 5′ adapter contaminants (without 3′ adapters or the insert tag), poly-A, T, G, or C, low-quality reads, and reads < 18 nt, produced clean sequencing reads. Then, Bowtie was used to map high-quality reads to a reference sequence, without mismatches, to analyze their expressions and distributions on the reference sequence. The RepeatMasker and Rfam databases were then used to remove tags originating from protein-coding genes, repeat sequences, rRNAs, transfer RNAs, small nuclear RNAs, and small nucleolar RNAs and sRNA tags. Comparisons were then made using clean reads of the miRNA precursors and the mature miRNAs from all animals in miRBase (version 22.0), with the goal of identifying each sample’s sequence and the number of miRNA families (not species-specific). Novel miRNAs were predicted by evaluating the characteristic hairpin structures of miRNA precursors.
For mRNA-seq, reads of a low-quality (> 20% of the bases qualities being < 10), with adaptors and with unknown bases (N bases > 5%), were removed to obtain clean reads. These were then assembled into unigenes, with unigene functional annotation, simple sequence repeat detection, and unigene expression level calculations. After differentially expressed genes (DEGs) were identified among the samples, a clustering analysis and functional annotations were performed. Raw reads were defined as reads of low-quality, adaptor-polluted, or having a high unknown base (N) content. Before the downstream analyses, internal software was used to filter these reads, which involved the removal of the following: (1) reads with adaptors; (2) reads with unknown bases (N) > 5%; and (3) low-quality reads (i.e., reads in which the percentage of bases with a quality < 10 was > 20%). After this filtering process, the remaining reads were termed “Clean Reads” and stored in FASTQ format. For de novo assembly, Trinity was used with the clean reads, and PCR duplication events were removed to improve the efficiency. Then, TIGR Gene Indices clustering tools were used to cluster transcripts to unigenes. Trinity contains three independent software modules: Inchworm, Chrysalis, and Butterfly. The sequence data was partitioned into many individual de Bruijn graphs, each indicating the transcriptional complexity at a given gene or locus. Each graph was then processed independently to extract full-length splicing isoforms. In addition, the system identified transcripts derived from paralogous genes.
miRNA and mRNA Differential Expression Analyses
To identify the differentially expressed miRNAs (DEMs) and mRNAs among the seven different stages, expression data were log2-transformed, and a scatter-plot was constructed. Briefly, the 21 libraries were used to normalize the miRNA expression data to obtain the number of transcripts per million reads (TPM), using the following normalization formula:
Normalized expression = mapped read count/total reads × 106.
The TPM analysis eliminated the influence of sequencing discrepancies in the miRNA expression calculations. The fold-change and p-values were calculated from the normalized expression and p-values, respectively, and then adjusted using the q-value. The criteria q-value < 0.01 and |log2(foldchange)|> 1 were chosen for determining significantly differential expression (default). In the analysis of mRNA expression abundance, the FPKM was used to indicate its expression level.
Transcriptome Annotation
Gene ontology (GO; www.geneontology.org) is an international standard classification system for gene functional determinations. The distribution of the selected target genes (miRNA and mRNA) among GO biological pathways/functions was used to characterize biological differences among samples on the basis of gene function. Using this method, the GO terms in the database (www.geneontology.org), as biological functions, were determined for candidate target genes, and the numbers of genes were calculated for every term. Significantly enriched GO terms were identified by performing a hypergeometric test on the target gene candidate list from the background reference gene list. The Kyoto Encyclopedia of Genes and Genomes (KEGG; www.kegg.jp) database is a public database containing biological pathway data, and it serves as a resource for discerning high-level functions and processes that are active in a biological system (Kanehisa et al. 2008). In this study, metabolic or signal transduction pathways, significantly enriched with target gene candidates, were identified using a KEGG pathway analysis and compared with a hypergeometric test and the background reference gene list.
Target Analysis of miRNAs and Development-Related Genes
Expression profiling identified plenty of DEGs with functions in larval development. However, the roles of genes in development were difficult to discern among the DEG pools. When analyzing larval development-relevant genes, GO terms and KEGG pathways were mainly those associated with development, including developmental processes (GO:0,032,502), post-embryonic development (GO:0,009,791), regulation of developmental processes (GO:0,050,793), regulation of cell development (GO:0,060,284), animal organ development (GO:0,048,513), cell development (GO:0,048,468), anatomical structure development (GO:0,048,856), central nervous system development (GO:0,007,417), multicellular organism development (GO:0,007,275), system development (GO:0,048,731), nervous system development (GO:0,007,399), regulation of growth (GO:0,040,008), germ cell development (GO:0,007,281), sensory system development (GO:0,048,880), developmental maturation (GO:0,021,700), cellular developmental process (GO:0,048,869), and tube development (GO:0,035,295). Seven adjacent comparisons were employed to identify 100 DEGs associated with development and identified 3′-UTRs used for miRNA-target prediction. To more accurately identify targets, multiple software programs were used. We used RNAhybrid (Jan and Marc 2006) and miRanda (John et al. 2005) to predict targets. For the RNAhybrid algorithm, the free energy (e) and p-value were used to estimate interactions between miRNAs and mRNAs. The criteria for functional targets were e < − 20 kcal/mol and p < 0.05. The miRNA functional target genes identified using the miRanda algorithm were restricted to those meeting the criteria score > 100 and free energy < − 10 kcal/mol.
Statistical Analysis
The R statistical software package (R, Auckland, New Zealand) was used to analyze the RNA-seq data. For graphical representations, R was used to correct for multiple testing and p-value corrections, and the data are expressed as the means ± standard errors, with p < 0.05 defining a significant difference. GraphPad 5.0 (San Diego, CA, USA) was used to generate figures.
Results
Small RNA Transcriptome Sequencing and Data Analysis
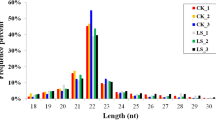
A preliminary analysis of the statistics from the 21 libraries was summarized in Table 1. Briefly, 21 small RNA libraries were established from triplicate samples of seven developmental stages using the BGISEQ-500 sequencing platform. On average, we obtained 24,476,982 raw tag counts per sample, and 22,073,637 (90.18% of the raw reads) clean counts after reads processing and filtering, with sequencing results having Q20 values above 98.50% and genome alignment ratios above 69%. Meanwhile, all clean reads were aligned with GenBank and Rfam databases and the unmapped reads were searched against miRBase database to identify known miRNAs. The composition of the RNA classes in each library is shown in Fig. 2. A total of 3,263,955 (14.79% of clean reads) and 4,659,534 (21.11% of clean reads) reads were obtained in mature and unmap counts, respectively. Length distribution analysis of the unique small RNA sequences showed that the majority of the unique small RNAs ranged from 21 to 23 nt in length, and the length of 22 nt was the most abundant (Fig. 3).

Annotation distribution of sRNAs. Clean reads annotated by aligning genome and blasting against the GenBank, Rfam, and miR-Base databases

Length distribution of sRNAs during seven different P. fucata development stages
Sequencing of miRNA from Different Developmental Stages
The number of known mature miRNA in each sample and the number of novel predicted miRNA are shown in Table 2. In total, 329 miRNAs, including 242 novel miRNAs, were obtained from the samples during the development of P. fucata. The libraries of adjacent developmental stages were compared. Briefly, 136 miRNAs were differentially expressed in the two Fer and Tro libraries, forming the largest differentially expressed group. Among them, 100 and 36 miRNAs were up- and downregulated respectively in fertilized eggs. Additionally, 72 miRNAs were differentially expressed in the two Fer and Tro libraries, with 26 and 46 miRNAs being up- and downregulated respectively in the egg stage.
Analysis of DEMs and Related Target Genes
Here, the miRNA expression level was calculated using TPM. Sample similarity analyses, including similarity, principal component, and correlation analyses, revealed the similarity and independence of sequencing samples (Suppl. Figs. 1 and 2). To screen for more accurate results, the combined free energy and score value were used to filter the data using miRanda and RNAhybrid software. After filtering, 255 targeted miRNAs and 8957 target genes, forming 15,430 targeting relationships, were obtained (Suppl. Fig. 3). The number of DEMs in each adjacent sample and their predicted target genes are shown in Suppl. Table 1. GO function classification statistics for the target genes of DEMs were developed, and the functional distribution characteristics of differentially expressed genes were investigated from a macro perspective (Suppl. Fig. 4). According to the predicted miRNA targeting patterns, there were five types: one miRNA regulated one target gene, one miRNA regulates multiple target genes, two or more miRNAs regulated multiple target genes, two or more miRNAs regulated a target gene, and combinations of two or more of the above targeting modes (Fig. 4).

Regulatory relationship of target genes. Note: Pentagram represents a miRNA, ellipse a target gene, red upregulated expression, and green downregulated expression
Sequencing of mRNA Transcriptomes and Data Analysis
To better understand the early developmental mechanisms in P. fucata larvae, the transcriptomes of samples from the seven stages were analyzed. Using mRNA-seq, the 21 cDNA libraries were constructed and sequenced on the DNBSEQ platform. A total of 93.3 gigabases (Gb) of clean data were obtained, and the Q20 was > 97.98%. Over 67.82% of the clean reads mapped to the pearl oyster genome (Table 3), and subsequently, 33,550 genes, including 26,333 known genes and 7217 predicted new genes, were identified through transcript assembly, based on the pearl oyster genome (Du et al. 2017). Furthermore, 59,897 new transcripts were detected, including 32,005 new alternative splicing subtypes of known protein-coding genes, 7335 transcripts belonging to new protein-coding genes, and 20,557 belonging to long-noncoding RNAs.
Identification and Functional Annotation of DEGs
Bowtie2 software was used to align clean reads to the pearl oyster genome, and they were then processed using RSEM software to define gene expression levels. Subsequently, DEGs between two adjacent samples were determined on the basis of gene expression levels using DEseq2 software. After normalization by FPKM, DEGs were identified between samples. The DEGs were displayed using MA plots with a color scheme corresponding to the methodology (Fig. 5). Genes regulated during metamorphosis were identified, and the number of upregulated genes (URGs) was significantly greater than downregulated genes (DRGs). In total, 10,149 DEGs were obtained in Fer vs Egg, including 5318 URGs and 4831 DRGs; 17,635 DEGs were obtained in from Tro vs Fer, including 10,344 URGs and 7291 DRGs; 15,253 DEGs were obtained in D-sha vs Tro, including 8624 URGs and 7867 DRGs; 12,410 DEGs were obtained in Umb vs D-sha, including 6053 URGs and 6357 DRGs; 14,184 DEGs were obtained in Ped vs Umb, including 7144 URGs and 7040 DRGs; and 16,491 DEGs were obtained in Met vs Ped, including 8624 URGs and 7867 DRGs (Fig. 6).

Differentially expressed genes between two adjacent groups displayed as MA plots. Gene expression values (mean of duplicate samples) shown in MA plot; upregulated genes in red, downregulated genes blue, and nondifferentially expressed genes in gray; and y-axis represents intensity ratio log2 (fold change) and x-axis average intensity

Statistics of differentially expressed genes
Functional Enrichment of DEGs
The functions of development-related DEGs were further determined by performing GO functional enrichment and KEGG pathway analyses. The GO enrichment analysis revealed that the gene functions in different samples were mainly concentrated in transmembrane signaling receptor, signaling receptor, molecular transducer activities, intrinsic components of membrane, integral components of membrane, membrane parts, and G protein–coupled receptor activity. The KEGG results showed that these DEGs were mainly involved in the regulation of neuroactive ligand-receptor interactions, Notch-signaling pathway, calcium-signaling pathway, cAMP-signaling pathway, and protein digestion and absorption (Suppl. Table 2).
Enrichment Analysis of DEM Target Genes and Expression Verification
Putative DEM targets were predicted using RNAhybrid and miRanda. A total of 8187 DEM target genes were predicted, and target gene functions were annotated by comparison with Nr, GO, and KEGG databases. Here, the target genes of the DEMs were mainly related to biological processes, such as biological regulation, localization, and cellular and metabolic processes. For cellular components, the identified genes were mainly related to membrane, membrane parts, cells, and organelles. For molecular function, genes were mainly related to binding, as well as to catalytic, transporter, and molecular transducer activities. To determine the enriched biological pathways, all the genes were explored using the KEGG database to categorize them into cellular processes, environmental information and genetic information processing, human disease, metabolism, and organismal systems. The top 14 pathways identified here were the pentose phosphate pathway, alanine, aspartate, and glutamate metabolism, inositol phosphate metabolism, lysosomes, TGF-beta-signaling pathway, purine metabolism, Hippo-signaling pathway, protein export, thermogenesis, glycosaminoglycan degradation, autophagy, protein processing in endoplasmic reticulum, Notch-signaling pathway, and thyroid hormone-signaling pathway (Fig. 7). To confirm the results of the small RNA-seq, the expression patterns of these mRNAs and miRNAs were analyzed, and the accuracy of the RNA-seq and miRNA-seq was verified using qRT-PCR. For this, eight DEGs and three DEMs related to larval development were selected for verification (Suppl. Table 3; Suppl. Fig. 5). The relative quantitative miRNA and mRNA expression levels and their sequencing expression trends were consistent.

Go and KEGG analysis of target genes in pearl oyster. GO functional classification of pearl oyster; x-axis indicates number of target genes and y-axis GO terms; and all GO terms grouped into three ontologies: blue, biological process; brown, cellular component: and orange, molecular function. KEGG pathway enrichment of known and novel DEMs. KEGG pathways for known and novel DEMs retrieved (http://www.kegg.jp/kegg/kegg1.html). Rich factor reflects proportion in a given pathway; circle areas represent relative numbers of genes in a pathway; and circle colors represent Q value range. (For interpretation of references to color, the reader is referred to the web version of this article.)
DEGs and DEMs Involved in the Regulation of Larval Development
The GO annotation identified 100 DEGs, including those encoding protein Wnt, Ras-like GTP-binding protein, tubulin alpha-1A chain, Pax-6, and apolipoprotein D, related to the regulation of larval developmental processes (Suppl. Table 4). A heat map of these 100 developmental genes, representing their transcriptional levels, was calculated using FPKM values of the samples (Fig. 8). Here, 95 DEMs, including miR-100-5p, miR-1986, miR-1994a, miR-34-5p_4, and miR-242a, identified by target gene GO annotation were involved in the regulation of larval development. The expression levels of these 95 candidate miRNAs during the different stages of P. fucata are shown in Suppl. Table 5. According to the expression levels of the two -omics, the correlation coefficients of the expression levels of these miRNAs and their target genes in different developmental stages were further analyzed, and targeting relationships with an absolute correlation coefficient value greater than 0.6 were screened. We determined targeting relationships having the same expression trends in 14 groups, with 9 and 5 groups showing positive and negative correlations, respectively (Fig. 9).

Differentially expressed profiles for mRNA associated with the development

Expression of development related miRNA–mRNA pair (|correlation coefficient|> 0.6)
Discussion
In the present study, we attempted to identify miRNA–mRNA pairs related to the development of P. fucata using transcriptomic data and small RNAs from seven developmental stages. A total of 93.6 Gb of mRNA sequences and 28.5 Gb of small RNA sequence were obtained. By using genomic sequences of P. fucata as a reference, 33,550 genes, including 7217 novel genes, and 329 miRNAs, including 242 novel miRNAs, were identified in these stages. A total of 100 DEGs and 95 DEMs were identified as being involved in the regulation of larval development. To date, miRNAs have been reported to play important roles in development, but miRNA–mRNA pairs related to development have not been reported in pearl oyster (Kocamis et al. 2013; Zhang et al. 2020; Wang et al. 2021a). Therefore, this investigation of the mRNA and miRNA expression profiles may help to reveal the molecular regulatory mechanisms of early developmental metamorphosis in shellfish and provide new reference data.
In this study, KEGG pathways containing DEGs from the whole developmental process were confirmed, and they mainly included neuroactive ligand-receptor interaction, Notch-signaling pathway, calcium-signaling pathway, cAMP-signaling pathway, and protein digestion and absorption, which revealed that DEGs play important roles in nervous system development, axis formation, muscle differentiation, and the digestive system. According to previous studies, cAMP is a key factor in the settlement and metamorphosis of P. fucata martensii (Zhang et al. 2021). Metamorphosis occurs through the early development of invertebrates. The morphology and living habits of larvae vary greatly among different developmental stages, which may lead to these genes having different roles. In addition, most DEGs and DEMs were found between the Fer and Tro developmental stages, and the number of URGs was much greater than DRGs. After fertilization, various physiological functions of organisms begin to start, and the biological processes involved are more complex, which may require more genes to participate. Consequently, there are more DEGs at this stage. Previous studies have also found that the number of URGs is greater than DRGs during the larval development of Sinonovacula constricta, but no specific reason has been provided (Niu et al. 2020).
A large number of miRNAs have been identified in vertebrates; however, research on miRNAs in marine invertebrates has been limited. Previous studies revealed that miRNAs are widely involved in physiological processes, including embryonic development (Niu et al. 2020; Wang et al. 2021a), biomineralization (Zhe et al. 2016; Huang et al. 2019; Peng et al. 2019), pigment deposition (Tian et al. 2018; Wang et al. 2021b), glucose metabolism (Sun et al. 2019), gonadal development (Tao et al. 2016; Huang et al. 2020), immune stimulation (Chen et al. 2016b; Tang et al. 2020), and the neuroendocrine system (Chen et al. 2016c). In the present study, the 15,430 identified miRNA–mRNA pairs were composed of 255 miRNAs and 8957 target genes. Bartel. (2009) found that each miRNA could have multiple target genes and that several miRNAs could also regulate the same target gene. Upon further investigation, we found that there were five miRNA–mRNA regulatory relationships and that most targeting relationships were positively correlated in the 15,430 miRNA–mRNA pairs (Fig. 4). This suggests that miRNAs not only inhibit, but also promote, mRNA expression through targeted binding.
Molecular mechanisms of oyster larval metamorphosis are studied a lot based on transcriptome, proteome, and metabolome at recent years. Foulon et al. (2019) revealed that 59 sequences are potentially involved in the adhesion of Crassostrea gigas larvae and that the von Willebrand Factor domain may play a role in cohesive adhesion. Zhang et al. (2021) integrated transcriptomics and metabolomics to identify the key metabolic mechanisms of P. fucata martensii larval metamorphosis. Xu and Zhang (2020) used transcriptomics and proteomics methods to examine the dynamics of genes and proteins during the larval settlement and metamorphosis of C. gigas. In this study, we identified 95 DEMs and 100 target genes involved in larval development by integrating miRNA-seq and mRNA-seq. We found that miR-31 is a potential target miRNA of pecanex-like protein 4 and GTPase genes. It is highly expressed in the trochophore stage and peaks during the Ped stage. Previous research also found that GTPase genes play important roles in neural development (Hae et al. 2003), and miR-31 is a potent regulator of vascular lineage-specific differentiation and development in vertebrates (Sun et al. 2009). In addition, miR-31 is involved in the formation of the embryonic sea urchin skeleton and plays crucial roles in development (Stepicheva and Song 2015). In addition, miR-184 regulates the development of female germ cells in Drosophila melanogaster (Wu et al. 2011). In this study, miR-184 expression was abundant, indicating that it plays important roles in larval development.
In addition, Pma_10003828 (notch1) was significantly upregulated from Tro stage to Umb stage and regulated by novel_mir106, novel_mir109, and novel_mir50. According to previous studies, the notch1 gene plays an important role in the notochord development of zebrafish, and the gene is strongly associated with body length, gonad development, and sex change in groupers (Renan et al. 2015; Yu et al. 2016). Therefore, we speculated that notch1 is likely to involve with the gonad development of P. fucata, which needs to be further verified. Yet, the three targeted miRNAs play a negative regulatory role on notch1, whether they contribute equally or not, and the mechanism they regulate the target gene is worth deep study. Moreover, we found that some gene-miRNA pairs, such as Pma_10019570 (CELA1)–novel_mir6, Pma_10013423 (bPRPs)–novel_mir73, and Pma_10020507 (ECMA)–novel_mir159, were significantly upregulated during the Ped stage, implying that they may be associated with the metamorphic development of larvae.
In the present study, some miRNA–mRNA pairs shared the same expression trend, some others had opposite expression trends. Here, miR-34_2 and Pma_10026543 (protocadherin Fat 4-like) pair had the same expression trend, whereas novel_mir161 and Pma_10005574 (Fzd4) pair had opposite expression trends. The absolute correlation coefficient value between the miRNA and mRNA expression level in each period was greater than 0.6. According to previous studies, protocadherin Fat 4-like is involved in the Wnt/PCP and the Hippo pathways in Drosophila, and it plays key roles in the homeostasis of mammalian tissues (Puppo et al. 2014). Fzd4 is a receptor that plays a key role in the Wnt/β-signaling pathway, and the activation of the Wnt/β-signaling pathway promotes cell proliferation, migration, and invasion (Kushwaha et al. 2020; Li et al. 2020a, b). Another study shows that Fzd4 regulates ventral blood vessel remodeling in the zebrafish retina (Caceres et al. 2019). In addition, Wnt family members regulate somatic axis development through nerves during embryonic development and are related to somite formation in Capitella capitate (Charron and Tessier-Lavigne 2005; Seaver and Kaneshige 2006). What functions these gene-miRNA pairs have in the early development of P. fucata is worthy of being further studied.
Conclusions
In this study, we integrated mRNA and small RNA sequencing from seven different stages of larval development; a comprehensive vision could be developed for understanding the miRNA function in this important biological process of P. fucata. This study provided miRNA expression profiles of P. fucata at seven important developmental stages for the first time. The results suggested that potential miRNA–mRNA pair regulated larval development and some miRNA–mRNA pairs with expression correlation. Extensive comparisons were performed using stage-specific miRNAs and significant DEMs, which provided important data for future research.
Data Availability
miRNAseq and RNAseq data have been deposited to the sequence read archive (SRA) with accession numbers SRR15315621–SRR15315641 and SRR15310576–SRR15310596, respectively. The datasets used and analyzed during the current study are also available from the corresponding author on reasonable request.
References
Addadi L, Joester D, Nudelman F, Weiner S (2010) Mollusk shell formation: a source of new concepts for understanding biomineralization processes. Chemistry 12(4):980–987. https://doi.org/10.1002/chem.200500980
Ambros V (2004) The functions of animal microRNAs. Nature 431:350–355
Baker SM, Mann R (1994) Description of metamorphic phases in the oyster Crassostrea virginica and effects of hypoxia on metamorphosis. Mar Ecol Prog Ser 104:91–99. https://doi.org/10.3354/meps104091
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215. https://doi.org/10.1016/j.cell.2009.01.002
Caceres L, Prykhozhij SV, Cairns E, Gjerde H, Duff NM, Collett K, Berman JN (2019) Frizzled 4 regulates ventral blood vessel remodeling in the zebrafish retina. Dev Dyn 248:1243–1256
Charron F, Tessier-Lavigne M (2005) Novel brain wiring functions for classical morphogens: a role as graded positional cues in axon guidance. Development 132:2251–2262
Chen CJ, Shikina S, Chen WJ, Chung YJ, Chiu YL, Bertrand J, Lee YH, Chang CF (2016a) A novel female-specific and sexual reproduction-associated dmrt gene discovered in the stony coral, Euphyllia ancora1. Biol Reprod 94(2):40, 1–13. https://doi.org/10.1095/biolreprod.115.133173
Chen H, Zhou Z, Wang H, Wang L, Wang W, Liu R, Qiu L, Song L (2016b) An invertebrate-specific and immune-responsive microRNA augments oyster haemocyte phagocytosis by targeting CgIκB2. Sci Rep 6:29591. https://doi.org/10.1038/srep29591
Chen H, Zhou Z, Wang L, Wang H, Liu R, Zhang H, Song L (2016c) An invertebrate-specific miRNA targeted the ancient cholinergic neuroendocrine system of oyster. Open Biol 6:160059. https://doi.org/10.1098/rsob.160059
Deng ZH, Wei HJ, Zhao W, Chen MQ, Yu G, Sun J, Li YN, Wang Y (2020) Embryonic development and larval cultivation of Paphia schnelliana (Dunker), a unique economic species of the Beibu Gulf. Aquaculture 533:736161. https://doi.org/10.1016/j.aquaculture.2020.736161
Du X, Fan G, Jiao Y, Zhang H, Guo X, Huang R, Zheng Z, Bian C, Deng Y, Wang Q, Wang Z, Liang X, Liang H, Shi C, Zhao X, Sun F, Hao R, Bai J, Liu J, Chen W, Liang J, Liu W, Xu Z, Shi Q, Xu X, Zhang G, Liu X (2017) The pearl oyster Pinctada fucata martensii genome and multi-omic analyses provide insights into biomineralization. GigaScience 6(8):1–12. https://doi.org/10.1093/gigascience/gix059
Felisa R, Gina MSN, Juan BP, Regina B (2019) Supply and larval traits at metamorphosis of a coastal marine invertebrate with a bi-phasic life cycle under contrasting oceanographic conditions. Prog Oceanogr 178:102201–102201
Foulon V, Boudry P, Artigaud S, Guérard F, Hellio C (2019) In silico analysis of Pacific Oyster (Crassostrea gigas) transcriptome over developmental stages reveals candidate genes for larval settlement. Int J Mol Sci 20:197. https://doi.org/10.3390/ijms20010197
Fujimura T, Wada K, Iwaki T (1995) Development and morphology of the pearl oyster larvae, Pinctada fucata. Venus 54:25–48
Hadfield MG, Meleshkevitch EA, Boudko DY (2000) The apical sensory organ of a gastropod veliger is a receptor for settlement cues. Biol Bull 198:67–76
Hae KJ, Hey-Kyoung L, Kogo T, Huganir RL (2003) The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J Neurosci 23:1119–1124
Huan P, Wang H, Dong B, Liu B (2012) Identification of differentially expressed proteins involved in the early larval development of the Pacific oyster Crassostrea gigas. J Proteomics 75:3855–3865
Huang S, Ichikawa Y, Yoshitake K, Kinoshita S, Igarashi Y, Omori F, Maeyama K, Nagai K, Watabe S, Asakawa S (2019) Identification and characterization of microRNAs and their predicted functions in biomineralization in the pearl oyster (Pinctada fucata). Biology 8:47. https://doi.org/10.3390/biology8020047
Huang T, Gu W, Liu E, Shi X, Wang B, Xu G, Yao Z (2020) Comprehensive analysis of miRNA-mRNA/lncRNA during gonadal development of triploid rainbow trout (Oncorhynchus mykiss). https://doi.org/10.21203/rs.3.rs-31357/v1
Jackson DJ, Ellemor N, Degnan BM (2005) Correlating gene expression with larval competence, and the effect of age and parentage on metamorphosis in the tropical abalone Haliotis asinina. Mar Biol 147:681–697. https://doi.org/10.1007/s00227-005-1603-z
Jackson DJ, Wörheide G, Degnan BM (2007) Dynamic expression of ancient and novel molluscan shell genes during ecological transitions. BMC Evol Biol 7:160. https://doi.org/10.1186/1471-2148-7-160
Jan K, Marc R (2006) RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res 34:451–461
Jiao Y, Zheng Z, Du XD, Wang QH, Huang RL, Deng YW, Shi SL, Zhao XX (2014) Identification and characterization of microRNAs in pearl oyster Pinctada martensii by solexa deep sequencing. Mar Biotechnol 16:54. https://doi.org/10.1007/s10126-013-9528-x
John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS (2005) MiRanda Application: Human microRNA targets. https://doi.org/10.1371/journal.pbio.0030264
Kanehisa M, Araki M, Goto S, Hattori M, Itoh M (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36:D480–D484
Kocamis H, Hossain MM, Cinar MU, Salilew-Wondim D, Mohammadi-Sangcheshmeh A, Tesfaye D, Hölker M, Schellander K (2013) Expression of microRNA and microRNA processing machinery genes during early quail (Coturnix japonica) embryo development. Poult Sci 92:787–797
Krol J, Loedige I, Filipowicz W (2010) The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11:597–610
Kushwaha P, Kim S, Foxa GE, Michalski MN, Williams BO, Tomlinson RE, Riddle RC (2020) Frizzled-4 is required for normal bone acquisition despite compensation by Frizzled-8. J Cell Physiol 235:6673–6683
Leise EM, Thavaradhara K, Durham NR, Turner BE (2001) Serotonin and nitric oxide regulate metamorphosis in the marine snailIlyanassa obsoleta. Am Zool 41:258–267. https://doi.org/10.1093/icb/41.2.258
Li HM, Liu BS, Huang GJ, Fan SG, Zhang B, Su JQ, Yu DH (2017) Characterization of transcriptome and identification of biomineralization genes in winged pearl oyster (Pteria penguin) mantle tissue. Comp Biochem Physiol Part D: Genomics Proteomics 21:67–76
Li HM, Bai LR, Dong XY, Qi XH, Liu HY, Yu DH (2020a) SEM observation of early shell formation and expression of biomineralization-related genes during larval development in the pearl oyster Pinctada fucata. Comp Biochem Physiol Part d: Genomics Proteomics 33:100650. https://doi.org/10.1016/j.cbd.2019.100650
Li ZT, Zhang X, Wang DW, Xu J, Kou KJ, Wang ZW, Sun XY (2020b) Overexpressed lncRNA GATA6-AS1 inhibits LNM and EMT via FZD4 through the Wnt/β-catenin signaling pathway in GC. Mol Ther -- Nucleic Acids 19:827–840
Li HM, Zhang B, Huang GJ, Liu BS, Fan SG, Zhang DL, Yu DH (2016) Differential gene expression during larval metamorphic development in the pearl oyster, Pinctada fucata, based on transcriptome analysis. Int J Genomics 2016:1–15
Liu G, Huan P, Liu BZ (2017) A SoxC gene related to larval shell development and co-expression analysis of different shell formation genes in early larvae of oyster. Dev Genes Evol 227(3):181–188. https://doi.org/10.1007/s00427-017-0579-2
Lopes KP, Vinasco-Sandoval T, Vialle RA, Paschoal FM, Bastos VAP, Bor-Seng-Shu E, Teixeira MJ, Yamada ES, Pinto P, Vidal AF, Ribeiro-Dos-Santos A, Moreira F, Santos S, Paschoal EHA, Ribeiro-Dos-Santos  (2018) Global miRNA expression profile reveals novel molecular players in aneurysmal subarachnoid haemorrhage. Sci Rep 8:8786. https://doi.org/10.1038/s41598-018-27078-w
Malumbres M (2012) miRNAs versus oncogenes: the power of social networking. Mol Syst Biol 8:569. https://doi.org/10.1038/msb.2012.2
Niu DH, Li BB, Xie SM, Dong ZG, Li JL (2020) Integrated mRNA and small RNA sequencing reveals regulatory expression of larval metamorphosis of the Razor Clam. Mar Biotechnol 22:696–705
Peng J, Zeng D, He P, Wei P, Hui W, Wu T, Lin Y (2019) mRNA and microRNA transcriptomics analyses in intermuscular bones of two carp species, rice flower carp (Cyprinus carpio var. Quanzhounensis) and Jian carp (Cyprinus carpio var. Jian). Comp Biochem Physiol Part d: Genomics Proteomics 30:71–80
Puppo F, Sebbagh M, Helmbacher F, Levy N, Krahn M, Bartoli M (2014) G.P.14 from muscular architecture to function: the involvement of FAT1 protocadherin in FSHD. Neuromuscular Disord 24:798. https://doi.org/10.1016/j.nmd.2014.06.028
Renan X, Trejo-Martinez J, Caballero-Arango D, Brule T (2015) Growth stanzas in an Epinephelidae–Lutjanidae complex: considerations to length-weight relationships. Rev Biol Trop 63:175–187
Seaver EC, Kaneshige LM (2006) Expression of ‘segmentation’ genes during larval and juvenile development in the polychaetes Capitella sp. I and H. Elegans. Dev Biol 289:179–194
Song H, Yu ZL, Sun LN, Gao Y, Zhang T, Wang HY (2016) De novo transcriptome sequencing and analysis of Rapana venosa from six different developmental stages using Hi-seq 2500. Comp Biochem Physiol Part D: Genomics Proteomics 17:48–57. https://doi.org/10.1016/j.cbd.2016.01.006
Stepicheva NA, Song JL (2015) MicroRNA-31 modulates skeletal patterning in the sea urchin embryo. Development 142:3769–3780
Sun F, Wang J, Pan Q, Yu Y, Zhang Y, Yang W, Ju W, Li X, An H (2009) Characterization of function and regulation of miR-24-1 and miR-31. Biochem Biophys Res Commun 380:660–665
Sun JL, Liu Q, Zhao LL, Cui C, Wu H, Liao L, Yang S (2019) Potential regulation by miRNAs on glucose metabolism in liver of common carp (Cyprinus carpio) at different temperatures. Comp Biochem Physiol Part D: Genomics Proteomics 32:100628. https://doi.org/10.1016/j.cbd.2019.100628
Suquet M, Labbé C, Puyo S, Mingant C, Quittet B, Boulais M, Queau I, Ratiskol D, Diss B, Haffray P (2014) Survival, growth and reproduction of cryopreserved larvae from a marine invertebrate, the Pacific Oyster (Crassostrea gigas). PloS One 9:e93486. https://doi.org/10.1371/journal.pone.0093486
Tang X, Tang J, Zhou Y, Qu F, Zou J, Liu XT, Z, (2020) Effects of sodium butyrate stimulation on immune-related mRNA-miRNA network in intestine of grass carp. Aquacult Res 52:309–322
Tao WJ, Sun LN, Shi HJ, Cheng YY, Jiang DN, Fu B, Wang D (2016) Integrated analysis of miRNA and mRNA expression profiles in tilapia gonads at an early stage of sex differentiation. BMC Genomics 17:328. https://doi.org/10.1186/s12864-016-2636-z
Teh CP, Zulfigar Y, Tan SH (2012) Epinephrine and l-DOPA promote larval settlement and metamorphosis of the tropical oyster, Crassostrea iredalei (Faustino, 1932): an oyster hatchery perspective. Aquaculture 338–341:260–263
Tian X, Pang X, Wang L, Li M, Dong C, Ma X, Li X (2018) Dynamic regulation of mRNA and miRNA associated with the developmental stages of skin pigmentation in Japanese ornamental carp. Gene 666:32–43
Vogeler S, Wikfors GH, Li X, Veilleux D, Joyce A (2019) Larval metamorphosis in four bivalve species in response to NMDA receptor ligands: the NMDA receptor pathway as potential regulator of bivalve transition to spat. Aquaculture 511:634173. https://doi.org/10.1016/j.aquaculture.2019.05.058
Wang L, Song F, Yin H, Zhu W, Fu J, Dong Z, Xu P (2021a) Comparative microRNAs expression profiles analysis during embryonic development of common carp, Cyprinus carpio. Comp Biochem Physiol Part d: Genomics Proteomics 37:100754. https://doi.org/10.1016/j.cbd.2020.100754
Wang P, Zeng D, Xiong G, Zhou X, Jiang H, Hu Y, Wang X (2021b) Integrated analysis of mRNA-seq and microRNA-seq depicts the potential roles of miRNA-mRNA networks in pigmentation of Chinese soft-shelled turtle (Pelodiscus sinensis). Aquacult Rep 20:100686. https://doi.org/10.1016/j.aqrep.2021.100686
Wei P, He P, Zhang X, Li W, Zhang L, Guan J, Chen X, Lin Y, Zhuo X, Li Q, Peng J (2019) Identification and characterization of microRNAs in the gonads of Crassostrea hongkongensis using high-throughput sequencing. Comp Biochem Physiol Part d: Genomics Proteomics 31:100606. https://doi.org/10.1016/j.cbd.2019.100606
Weiss IM, Tuross N, Addadi L, Weiner S (2010) Mollusc larval shell formation: amorphous calcium carbonate is a precursor phase for aragonite. J Exp Zool 293:478–491
Wienholds E, Kloosterman W, Miska E, Alvarez-Saavedra E, Berezikov E, Bruijn ED, Horvitz HR, Kauppinen S, Plasterk RHA (2005) MicroRNA expression in zebrafish embryonic development. Science 309:310–311
Wu J, Bao J, Wang L, Hu Y, Chen X (2011) MicroRNA-184 downregulates nuclear receptor corepressor 2 in mouse spermatogenesis. BMC Dev Biol 11:1–10
Xu F, Wang XT, Feng Y, Huang W, Wang W, Li L, Fang XD, Que HY, Zhang GF (2014) Identification of conserved and novel microRNAs in the Pacific oyster Crassostrea gigas by deep sequencing. PLoS One 9:e104371. https://doi.org/10.1371/journal.pone.0104371
Xu F, Zhang GF (2020) Transcriptomic and proteomic dynamics during metamorphosis of Pacific oyster Crassostrea gigas. https://doi.org/10.1101/2020.03.25.004614
Xue W, Wang WJ, Li Z, Sun GH, Xu XH, Feng YW, Luo QH, Li B, Zhang Q, Yang J (2021) Comprehensive analysis of differentially expressed ncRNA, mRNA, and their ceRNA networks in the regulation of glycogen content in the Pacific oyster Crassostrea gigas. Aquaculture 531:735895. https://doi.org/10.1016/j.aquaculture.2020.735895
Yu DL, Wu HF, Peng X, Ji CL, Zhang XY, Song J, Qu JL (2020) Profiling of microRNAs and mRNAs in marine mussel Mytilus galloprovincialis. Comp Biochem Physiol Part c: Toxicol Pharmacol 230:108697. https://doi.org/10.1016/j.cbpc.2019.108697
Yu H, You XX, Li J, Liu HK, Meng ZN, Xiao L, Zhang HF, Lin HR, Zhang Y, Shi Q (2016) Genome-wide mapping of growth-related quantitative trait loci in orange-spotted grouper (Epinephelus coioides) using double digest restriction-site associated DNA sequencing (ddradseq). Int J Mol Sci 17:501. https://doi.org/10.3390/ijms17040501
Yu J, Zhe Z, Tian R, Du X, Wang Q, Huang R (2015) MicroRNA, Pm-miR-2305, participates in nacre formation by targeting pearlin in pearl oyster Pinctada martensii. Int J Mol Sci 16:21442–21453
Zhang GF, Fang XD, Guo XM, Li L, Luo RB, Xu F, Yang PC, Zhang LL, Wang X, Qi H, Xiong Z, Que H, Xie Y, Holland PWH, Paps J, Zhu Y, Wu F, Chen Y, Wang J, Peng C, Meng J, Yang L, Liu J, Wen B, Zhang N, Huang Z, Zhu Q, Feng Y, Mount A, Hedgecock D, Xu Z, Liu Y, Domazet-Lošo T, Du Y, Sun X, Zhang S, Liu B, Cheng P, Jiang X, Li J, Fan D, Wang W, Fu W, Wang T, Wang B, Zhang J, Peng Z, Li Y, Li N, Wang J, Chen M, He Y, Tan F, Song X, Zheng Q, Huang R, Yang H, Du X, Chen L, Yang M, Gaffney PM, Wang S, Luo L, She Z, Ming Y, Huang W, Zhang S, Huang B, Zhang Y, Qu T, Ni P, Miao G, Wang J, Wang Q, Steinberg CEW, Wang H, Li N, Qian L, Zhang G, Li Y, Yang H, Liu X, Wang J, Yin Y, Wang J (2012) The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490:49–54
Zhang J, Xiong X, Deng Y, Zheng Z, Yang C, Du X (2021) Integrated application of transcriptomics and metabolomics provides insights into the larval metamorphosis of pearl oyster (Pinctada fucata martensii). Aquaculture 532:736067. https://doi.org/10.1016/j.aquaculture.2020.736067
Zhang Q, Dou W, Song ZH, Jin TJ, Yuan GR, De Schutter K, Smagghe G, Wang JJ (2020) Identification and profiling of Bactrocera dorsalis microRNAs and their potential roles in regulating the developmental transitions of egg hatching, molting, pupation and adult eclosion. Insect Biochem Mol Biol 127:103475. https://doi.org/10.1016/j.ibmb.2020.103475
Zhe Z, Jiao Y, Du XD, Tian Q, Wang Q, Huang R, Deng Y (2016) Computational prediction of candidate miRNAs and their potential functions in biomineralization in pearl oyster Pinctada martensii. Saudi J Biol Sci 23:372–378
Zheng Z, Huang RL, Tian RR, Jiao Y, Du XD (2016) Pm-miR-133 hosting in one potential lncRNA regulates RhoA expression in pearl oyster Pinctada martensii. Gene 591:484–489
Funding
This work was supported by the National Key R&D Program of China (2018YFD0901406), the National Natural Science Foundation of China (31873042), the Guangxi Key R&D Program (2018AB52002), the Guangxi Natural Science Foundation Program (2021GXNSFAA075008), the and Guangxi Key Laboratory of Marine Biodiversity Conservation Program (2021ZB02).
Author information
Authors and Affiliations
Contributions
Jian Chen and Dahui Yu contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Jian Chen, Ziqin Zhai, Lili Lu, Suping Li, and Lirong Bai. The first draft of the manuscript was written by Jian Chen, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, J., Zhai, Z., Lu, L. et al. Identification and Characterization of miRNAs and Their Predicted mRNAs in the Larval Development of Pearl Oyster Pinctada fucata. Mar Biotechnol 24, 303–319 (2022). https://doi.org/10.1007/s10126-022-10105-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-022-10105-3