
Abstract
The razor clam, Sinonovacula constricta, is an important economic marine shellfish, and its larval development involves obvious morphological and physiological changes. MicroRNA plays a key role in the physiological changes of the organism through regulating targeted mRNA. This study performed miRNA–mRNA sequencing for eight different developmental stages of S. constricta using Illumina sequencing. A total of 2156 miRNAs were obtained, including 2069 known miRNAs and 87 novel miRNAs. In addition, target genes were predicted for key miRNAs differentially expressed between adjacent development samples by integrating the mRNA transcriptome. Further analysis revealed that the differentially expressed genes were enriched in complement activation, alternative pathways, translation, and negative regulation of monocyte molecular protein-1 production. KEGG pathway annotation showed significant enrichment in the regulation of the ribosome, phagosome, tuberculosis and fluid shear stress, and atherosclerosis. Ten mRNAs and ten miRNAs that are related to larval metamorphosis were identified using real-time PCR. Furthermore, the double luciferase experiment validated the negative regulatory relationship between miR-133 and peroxisome proliferator-activated receptor-γ (PPAR-γ). These results indicated that the target genes regulated by these differentially expressed miRNAs may play an important regulatory role in the metamorphosis development of S. constricta.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The early larval development of shellfish is a major feature that affects individual survival rate, population number (Vogeler et al. 2019), spatial distribution, and resource supplementation (Rey et al. 2019). Thus, the genetics and the regulatory mechanisms of larval metamorphosis in shellfish have been extensively investigated. Genetic and environmental factors such as gene expression (Okazaki and Shizuri 2000), genomic variation (Henry et al. 2010), hormonal regulation (Applebaum et al. 2012), nutrients (Weaver et al. 2018), light (Weaver et al. 2018), water (McCormick and Molony 1995), and temperature (O’Connor et al. 2015) control the development of the embryo and larva of bilateral animals. Attachment metamorphosis of shellfish seedlings is a key period in their life history (Sun et al. 2020). There are several factors affecting shellfish during the metamorphosis process that could explain the relatively low post-larvae yield observed in hatcheries (Tremblay et al. 2020). The occurrence of shellfish metamorphosis is related to many factors, such as the genetic characteristics, age, and nutritional status of larvae (Zheng et al. 2019). At the same time, it is also affected by external environmental physical factors (Hettinger et al. 2012), chemical factors (Garcia-Lavandeira et al. 2005), and biological factors (Campbell et al. 2011). The razor clam, S. constricta, is an important commercial species in China. As one of the typical coastal shellfish, the clam has important economic and nutritional value (Niu et al. 2016). Similar to most marine bivalves, the larvae of S. constricta have a transition from plankton to benthic life (Lukwambe et al. 2019). Larval metamorphosis is an essential developmental stage in the life of S. constricta, and both morphological and physiological metamorphoses involve remarkable changes. Therefore, under the influence of various factors, it is an important research direction to understand the specific molecular regulation mechanism of larval metamorphosis.
Although there are many factors affecting the conversion of larvae to adults of shellfish, the related molecular mechanisms are still poorly understood. Neurotransmitters that affect G protein-coupled receptors (GPCRs) are involved in larval metamorphosis in many species, including insects (Kang et al. 2019), amphibians (Conaco et al. 2012), fish (Parichy et al. 2003), and barnacles (Okazaki and Shizuri 2000). In marine bivalves, GPCRs and signal transduction by AC/cAMP pathway could mediate the metamorphosis of larvae in the mussel Mytilus edulis (Liang et al. 2018). Transcriptome sequencing facilitated the study of molecular mechanisms. Recently, studies revealed that the biomineralization genes and repair processes were related to shell formation during the larval development of the pearl oyster Pinctada fucata larvae by transcriptome, and found that the Wnt pathway played an important role (Li et al. 2020a). In the transcriptome analysis of the Portuguese oyster Crassostrea angulata larvae, receptor of activated C kinase 1 (RACK1) was isolated and characterized, which played an important role in tissue differentiation or cell growth during larval metamorphosis of the oyster (Yang et al. 2014).
MicroRNA is a regulatory small molecule that widely participates in the regulation of biological growth and development, immunity, metabolic pathway and cell differentiation, and other life processes (Kim et al. 2009). Generally, miRNAs mainly regulate growth and development through post-transcriptional inhibition of target genes. A miRNA may regulate multiple target mRNAs, and one gene may also be regulated by multiple miRNAs (Bartel 2009). To date, at least 38,589 hairpin precursors and 48,860 mature microRNAs have been identified in 271 organisms (Kozomara et al. 2019). The results of miRNA sequencing on oysters indicated that some miRNAs may play an important role in organ development. For example, cgi-miR-1990-3p was specifically expressed in the mantle, cgi-miR-1991-5p was specifically expressed in the adductor muscle, and some miRNAs were expressed in large quantities in the stage of creeping larvae, such as cgi-miR-33-5p. The miRNAs of hsa-miR-33a-5p and hsa-miR-33b-5p mainly regulated insulin signaling pathway and fatty acid metabolism (Xu et al. 2014). However, the precise regulatory mechanisms and biological functions of many miRNAs are not clear. So, it is of great significance to elucidate the specific molecular mechanism of miRNAs in the metamorphosis of S. constricta.
In a previous study, we found that genes related to cellular process and macromolecule metabolism pathways played an important role in larvae metamorphosis of S. constricta (Niu et al. 2016). In order to understand how relevant functional genes are regulated during the metamorphosis period, we further conducted joint sequencing analysis of mRNA and miRNA at the same development time and obtain the regulatory relationship between mRNA and miRNA during the critical development stages. Through gene annotation and pathway analysis of target mRNAs regulated by differentially expressed miRNAs, it is intuitively shown that the regulatory effects of key miRNAs and genes affect the development of larval metamorphosis. The combined transcriptome data would further fill the gaps in miRNA research in S. constricta. In addition, it could provide a reference for the molecular mechanisms involved in larval development of marine bivalves.
Materials and Methods
Experimental Animals and Sample Preparation
The production of razor clams was performed in the Donghang Breeding Base in Zhejiang Province, China. The healthy adult clams were selected and placed in net cages with a volume of 1 × 0.5 × 0.2 m3, which were suspended in the breeding ponds. A shade net covered the cages to create a dark environment in order to stimulate spawning.
Sample Collection and RNA Extraction
According to the development time after fertilization, combined with microscope observation, the different developmental stages were determined (Qi and Yang 1984). The samples were collected in the cleavage period (Cp), trochophore stage (T), veliger stage (V), umbo larvae (U), creeping larvae (C), single pipe larvae (S), and juvenile (J) at 2–3 h, 4–5 h, 6 h, 1 day, 2–3 days, 5–6 days, 7–10 days, and 18 days after fertilization, respectively. Eight samples from eight development stages were sequenced, respectively. Additionally, the larvae were very small, less than 250 μm. So, each sample contained thousands of individuals. Total RNA was extracted from each sample by TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer’s recommendations. The values of OD260/280 and OD260/230 of total RNAs were measured by the NanoDrop 2000/2000c (Thermo, USA) to ensure the quality of the extracted total RNAs. The integrity of the total RNAs was verified by agarose gel electrophoresis to ensure the successful construction of the sequencing library.
HiSeq Sequencing and Data Analysis of mRNA
The mRNA transcriptome sequencing was performed on the Illumina HiSeq 2000 platform to generate pair-end fragments. The fragments were purified, and the sequences included in the sequencer and small fragments with low quality were removed. The program Trinity (http://trinityrnaseq.sourceforge.net/) was used to get the transcript sequences (Grabherr et al. 2013). The TGICL software was used to remove redundant extensions to obtain a final set of unigenes (Geo et al. 2003). According to the similarity and length of sequences, the longest one was selected as the unigene. Quality control was carried out for each sample. In the analysis of mRNA expression abundance, the FPKM was used to indicate its expression level. Due to the large number of samples, this study performed multi-sample statistics, PCA analysis, and cluster analysis between samples. The expression level of genes was calculated according to the negative binomial distribution test in DESeq software. Negative binomial distribution test was used to test the significance of the difference in reads.
HiSeq Sequencing and Data Analysis of Small RNA
Total RNA was extracted from the same samples for electrophoresis. The recovered 18–30 nt RNAs were ligated to the adapters of Illumina by T4 ligase. The ligation products were extracted by polyacrylamide denaturing electrophoresis. After reverse transcription and amplification, the amplified products were small RNA transcriptome sequencing libraries. The constructed library was qualified by HiSeq2000 and then sequenced. The adapter sequences in raw reads were removed to obtain small RNA sequences of varying lengths. Through quality control, we removed the base sequence with too low ratio of Q20 and got the clean reads for subsequent blast and annotation. In order to classify and annotate the small RNA, the clean reads were compared and annotated with the Rfam database, the cDNA sequence, the species repeat library, and the miRBase database in order. Novel miRNAs were predicted by mireap v0.2 (https://sourceforge.net/projects/mireap/) (Friedlaender et al. 2012). The expression abundance of miRNA was calculated using TPM. The abundance reflected the miRNA expression level (Sun et al. 2014).
Target Gene Prediction and Gene Annotation
Prediction of the target genes of differentially expressed miRNAs in adjacent samples was conducted using the Miranda software packages (John et al. 2004). Gene function was annotated based on the following databases: nonredundant protein sequence database (NR), a manually annotated and reviewed protein sequence database (SWISSPROT), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG). The KEGG annotation information of unigenes was obtained using KAAs (http://www.genome.jp/kaas-bin/kaas_main). Based on the annotation results of SwissProt, the GO term was mapped according to the UniProt ID, and then, the GO protein functional annotation information of unigenes was obtained.
Verification of MiRNA and mRNA Expression
Reverse transcription quantitative real-time PCR (qRT-PCR) was used to verify the sequencing data by CEX96™ Real-time System (Bio-Rad, USA). In this study, ten miRNAs, namely miR-133, miR-10a-5p, miR-317, miR-21, miR-184-3p, miR-981, aae-miR-92b-3p, lmi-miR-281-3p, lgi-miR-1985, and lgi-miR-2d and ten mRNAs, namely PPAR-γ, death-associated protein kinase (DAPK), ras-related C3 botulinum toxin substrate 1 (RAC1), ryanodine receptor (RyR2), inositol oxygenase (IO), adenylate kinase (ADK), ras-like GTP-binding protein (RLBP), rho GTPase-activating protein (RhoGAP), protein wnt-4 (Wnt4), and cyclic guanosine monophosphate (cGMP) were selected for verification using qRT-PCR in triplicate. They were expressed differentially in larval development. Total RNA was extracted from samples in different developmental stages of S. constricta using TRIzol reagent (Invitrogen, CA, USA) and converted into cDNA of miRNA and mRNA using miScript II RT Kit (Qiagen, Germany). MiScript SYBR Green PCR Kit (Qiagen, Germany) was used to perform qRT-PCR according to the manufacturer’s instructions. The primers for the reaction are shown in Supplementary Table 1. The expression was analyzed using the method 2–∆∆Ct.
Validation of the Targeting Relationship Between miR-133 and PPAR-γ
In the quantitative analysis of miRNAs and mRNAs, we detected the targeting relationship between miR-133 and PPAR-γ. To construct the PPAR-γ vectors, the ORF of PPAR-γ was amplified using the PPAR-γ-F1-SacI/JNK-R1-XbaI primers. The PCR products and pmirGLO luciferase reporter vector (Promega, USA) were treated with the corresponding restriction enzyme, and the ORF was inserted into the vector to obtain pmirGLO–PPAR-γ. The recombinant plasmid was validated by sequencing.
The Hek293T cells in the logarithmic growth phase were transferred into 24-well plates prior to the experiment and allowed to grow to approximately 80% confluence. The cells were washed with DPBS (Thermo, USA) and incubated with an antibiotic-free nutrient solution. The plasmid was transfected using Lipofectamine 3000 reagent (Invitrogen, USA), according to the manufacturer’s instructions. The control group was treated with the same volume of the negative plasmid. Each experiment was performed in triplicate.
For the identification of the miRNA targets, 293T cells were transfected with PPAR-γ–3′UTR reporter vector, together with miR-133 mimics, miR-NC, and control conditions. Three parallel experiments were set for each group. Reporter luciferase activity was measured with a Dual-Luciferase Reporter assay system (Promega, USA). Specifically, 2 mL of 5 × PLB was mixed with 8 mL of sterile water and stored at 4 °C. Then, 2.1 mL of 50 × Stop&Glo® substrate was mixed with 105 mL of Stop&Glo® buffer, dispensed into 2 mL brown enzyme-free frozen storage tubes, and stored at − 20 °C. The cell culture medium in the 24-well cell culture plate was removed and the cells were washed twice with DPBS. Next, 100 μL of passive lysate PLB was added to each well and shaken at room temperature for 15 min, before the lysate was collected in a 1.5-mL enzyme-free centrifuge tube. To detect firefly luciferase activity, 100 μL of LAR II and 20 μL of the previous lysate were mixed and measured after adding 100 μL of the Stop&Glo reagent. Relative luciferase activity values were obtained using firefly luciferase activity/renilla luciferase activity.
Results
Annotation of mRNA from Development Stages
In this study, 250,000 pairs of reads were randomly selected from the sequencing sample data, compared with the NR database to get clean reads. All the clean reads were used to perform de novo assembly, and a total of 77,833 unigenes were generated. The total number of bases of unigenes was 96,166,860 bp, where the max length of unigenes was 28,419 bp, the min length of unigenes was 301 bp, the average length of unigenes was about 1236 bp, and N50 was 2032 bp.
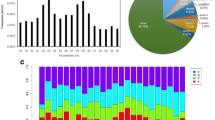
By comparing with multiple databases, the intersection and union annotation results of different databases were obtained (Fig. 1). The results showed that there were 23,611 annotated unigenes, accounting for about 30.34%. There were 6956 unigenes that can be blasted and annotated in all databases, accounting for 8.94%. The gene expression was calculated by the FPKM method.

Intersection and union graphs of NR, SwissProt, KOG, GO, and KEGG annotations in unigenes
Differentially Expressed Genes in Different Development Stages
After conducting principal component analysis on multiple samples, the results showed a distinct position of different developmental stages. After normalization by FPKM, we obtained differentially expressed genes between samples (Fig. 2). It showed the regulated gene during the process of metamorphosis, and the number of upregulated genes was significantly more than the number of downregulated genes. Between the J and V, 3036 genes were upregulated and 1000 genes were downregulated. It showed that more genes were differentially expressed in larvae from veliger to juvenile stages. Besides, in the comparisons between J and T, S and T, and S and V, 3667, 3373, and 3262 differentially expressed genes were identified, respectively.

Analysis of differentially expressed genes in different development stages. a Two-component principal component analysis of transcriptomes from eight developmental stages. The closer the PCA distance, the more similar the sample. b Statistics of differentially expressed transcripts. The conditions for screening differences are that p value is less than 0.05 and the difference multiple is greater than 2
Functional Enrichment of Differentially Expressed Genes
To further describe the function of differentially expressed genes, we performed GO function enrichment and KEGG pathway analysis on the differentially expressed genes screened during development. GO enrichment analysis of differentially expressed genes showed that the gene functions with different samples were mainly concentrated in biological functions such as complement activation, alternative pathways, translation, and negative regulation of monocyte molecular protein-1 production. The analysis results of KEGG showed that genes differentially expressed during development were mainly involved in the regulation of ribosome, phagosome, tuberculosis and fluid shear stress, and atherosclerosis.
Sequencing of MiRNA in Different Developmental Stages
The small RNA libraries were sequenced, and a total of 152,491,312 clean reads were generated. After comparing and filtering with the reference genome, Rfam database, transcript sequence, repeat sequence, miRbase database, etc., the sequence annotation was performed and novel miRNAs were predicted. The number of known mature miRNAs in each sample and the number of novel predicted miRNAs are shown in Table 1. A total of 2156 miRNAs were obtained from the samples during the development of S. constricta, including 2069 known miRNAs and 87 novel miRNAs. The known and novel miRNAs in the Cp were 1145 and 53, respectively; the Bp was 483 and 66, respectively; the T was 798 and 69; the V was 742 and 67; the U was 735 and 59,; the C was 624 and 37; the S was 772 and 47; and the J was 1110 and 46, respectively.
Analysis of Differentially Expressed MiRNA and Related Target Genes
In this study, TPM was used to calculate the expression level of miRNAs. Analysis of the similarity of samples including similarity test, principal component analysis, and cluster analysis showed the similarity and independence of sequencing samples (Supplementary Fig. 1). After differential screening statistics, we obtained 1179 miRNAs with differential expression pattern and 68 miRNAs were expressed with a high level (> 50 TPM). We counted the differentially expressed miRNAs during different development stages and predicted target genes of differentially expressed miRNAs in adjacent samples by using the Miranda software (John et al. 2004). The number of differentially expressed miRNAs in each adjacent sample and their predicted target genes are shown in Supplementary Table 2. It can be seen that one miRNA can regulate multiple target mRNAs, and one target mRNA can also be regulated by multiple target genes.
Verification of MiRNA and mRNA Expression in Development Stages
As found in the present analysis, the target genes of differentially expressed miRNAs mainly were related with biological processes such as DNA transcription, cell division, cell differentiation, and signal transduction. It involved some molecules that may play a role in ATP binding, metal ion binding, DNA and RNA binding, and silk/threonine kinase activation in cell components such as the nucleus, cytoplasm, mitochondria, and extracellular matrix (Supplementary Table 3). Besides, the results of KEGG analysis indicated that the target genes of differentially expressed miRNAs were mainly involved in 68 KEGG signaling pathways, including MAPK, TNF, Jak-STAT, Relaxin, FOXO, and other signaling pathways (Supplementary Table 4).
To confirm the results of small RNA-seq, we analyzed the expression patterns of the mRNAs and miRNAs and verified the accuracy of the RNA-seq and miRNA-seq using qRT-PCR. Ten DEGs and ten DE miRNAs related to larval metamorphosis were selected for verification (Fig. 3). The results showed that the relative quantitative expression levels of miRNA and mRNA and their sequencing expression trends were basically consistent.

Comparison of expression values of ten mRNAs and ten miRNAs in the different developmental stages of S. constricta (Cp, cleavage period; Bp, blastula period; T, trochophore stage; V, veliger stage; U, umbo larvae; C, creeping larvae; S, single pipe larvae; J, juvenile stage) by RNA-seq and qRT-PCR analyses. a The RNA-seq and qRT-PCR analyses of mRNAs. Histograms represent the relative expression levels (Log2) (FPKM) as assessed by RNA-seq; black dotted lines represent the expression levels of the relative expression by qRT-PCR and reported as means and standard errors of three biological replicates for each treatment. b The miRNA-seq and qRT-PCR analyses of miRNAs. Histograms represent the relative expression levels (Log2) (TPM) as assessed by miRNA-seq; black dotted lines represent expression levels of the relative expression by qRT-PCR and reported as means and standard errors of three biological replicates for each treatment
The Targeting Relationship Between mRNA and MiRNA
As shown in the electrophoresis results, we constructed the pmirGLO–PPAR-γ vector consistent with the target fragment and verified this result by sequencing (Fig. 4a). As shown in Fig. 4b, the results of the luciferase activity test showed that miR-133 inhibited the expression of PPAR-γ compared with the control group and the NC group (p < 0.01).

Verification of the targeting relationship between miR-133 and PPAR-γ. a Identification of luciferase reporter carrier by SacI/XbaI double enzyme electrophoresis (D SacI/XbaI double enzyme pmirGLO–PPAR-γ cloning; M, marker). b Relative luciferase activity of Hek293T cells in each groups/%. Control: Hek293T cells transfected with pmirGLO–PPAR-γ–3′UTR, miR-133 mimics: Hek293T cells co-transfected with pmirGLO–PPAR-γ–3′UTR and miR-133 mimics, miR-NC: Hek293T cells co-transfected with pmirGLO–PPAR-γ–3′UTR and miR-NC. **p < 0.01 vs control
Discussion
Metamorphosis is a typical biological phenomenon in the growth and development stages of many invertebrates (Clare et al. 1998). The development of shellfish larvae undergoes changes in organ formation and life habits, so it is a very important period and one of the important factors affecting the success rate of seedling reproduction (Huan et al. 2012). In previous studies, the research on the metamorphosis of bivalve was involved in the exploration period, and most of them were involved in neurotransmitters such as serotonin, catecholamine (Yang et al. 2013), L-DOPA (Satuito et al. 1999), and GABA (Sanchez-Lazo et al. 2012). However, the research on the biology of S. constricta is at the preliminary stage, especially the research on larval metamorphosis development. Currently, it has been shown that dopamine beta-hydroxylase plays an important regulatory role in larval metamorphosis and juvenile growth (Li et al. 2020b).
In order to understand the role of the miRNA–mRNA network in this important process, this study used a new generation of sequencing methods to conduct integrated sequencing research on mRNA and miRNA in different development stages. This study revealed the molecular regulatory mechanism of early developmental metamorphosis of shellfish from the perspective of joint analysis and provided some reference information. Next-generation sequencing technology was used to analyze the transcriptome changes in eight different metamorphic developmental stages of S. constricta. The de novo assembly generated 77,833 unigenes. The functional annotation of the differentially expressed genes showed that the differentially expressed genes were involved in the metamorphosis and development of S. constricta. This pioneering study provides us with a molecular basis for early metamorphic development in post-transcriptional regulation.
The basis of morphological changes in metamorphic development is cell division, differentiation, and apoptosis (Su et al. 1998). GO and KEGG analysis of the differentially expressed targeted genes in adjacent samples revealed that multiple molecular processes and life activities were involved in the process of metamorphosis (Peng et al. 2020), including DNA- and RNA-related activities, cell replication, division and differentiation, and participation in known signaling pathways related to development, growth, and immunity.
MiRNA, as a class of small noncoding RNA with a size of about 22 bp, is one of the hot spots in molecular biology research. In this study, a total of 2156 miRNAs were obtained by small RNA sequencing from samples in larval metamorphosis, including 2069 known miRNAs and 87 newly predicted miRNAs, of which 1179 miRNAs were differentially expressed. We obtained the genes targeted by predicting target genes for differentially expressed miRNAs. We found that the target genes predicted by differentially expressed miRNAs were mainly enriched in MAPK, TNF, Jak-STAT, Relaxin, FOXO, and other signaling pathways. Thyroid hormone (TH) is a hormone secreted by the thyroid gland. It is an amino acid derivative that has a wide range of functions in individual development (Brown et al. 2014), maintaining internal environment stability (Scholz et al. 2012), cell proliferation, and differentiation (Wang et al. 2011). Studies have found that the thyroid hormone can accelerate the bone formation of sea urchin mesenchymal cells through extracellular signal-regulated kinase 1/2-mediated integrin membrane receptors in the MAPK signaling pathway, thereby promoting bone development in larval sea urchins (Elias and Andreas 2018). In the present study, it was found that MAPK signals can mediate gradual metamorphosis of worms with bacterial inducers (Shikuma et al. 2016). In this study, we found that miR-2d differentially regulated cGMP expression. cGMP is a class of cyclic nucleotides that can be activated by protein kinases activated by G protein–linked receptors and then transduce extracellular signals to the nucleus (Kalia 2013). At present, studies have shown that cGMP signals mediate larval attachment and metamorphosis, such as the barnacle Balanus amphitrite (Zhang et al. 2012), the sea slug Alderia willowi (Romero et al. 2012), the bryozoan Bugula neritina (Yang et al. 2018), etc. This provides new ideas for subsequent related research in S. constricta.
The regulatory relationship between miRNAs and genes includes positive and negative regulation and, in most cases, negative regulation (Yu and Kuo 2019). Whether the target genes were negatively correlated, we determined the regulatory relationship of a pair of targeted relationships by constructing a vector and performing a double luciferase experiment. PPAR-γ is one of the subtypes of the peroxisome proliferator-activated receptor family and is an important cell differentiation transcription factor. A report presented evidence that PPARs can act as a coregulatory factor for TR in the thyroid signaling pathway (Bogazzi et al. 1994). At present, many studies have shown that PPAR-γ is involved in the differentiation of adipocytes during development (Schoonjans et al. 1996). Whether PPAR-γ is indirectly involved in regulating the process of metamorphosis is unclear at present and awaits further study. In this study, pmirGLO–PPAR-γ was successfully constructed by double-digesting the PPAR-γ target site and the pmirGLO vector, and a double luciferase verification experiment was performed. The results showed that miR-133 negatively regulated PPAR-γ. It provides data support for studying the further targeting relationship between miRNA and target genes and lays a foundation for the follow-up research on the regulation mechanism of PPAR-γ on the growth and metamorphosis development of S. constricta.
Conclusion
With the integrated data from mRNA and small RNA sequencing in different stages of early metamorphosis, a comprehensive vision could be developed for understanding the miRNA function in this important biological process of S. constricta. In this study, we firstly conducted the integrated analysis of mRNA and miRNA transcriptomes from eight different early developmental stages of S. constricta. The results suggested that potential miRNA–mRNA pairs involved in cell division, cell differentiation, and signal transduction regulated larval metamorphosis of S. constricta. Fluorescence quantification was used to initially verify the accuracy of the combined sequencing. The results of the Dual-Luciferase experiment based on miR-133 and PPAR-γ also further verified the regulation of miRNA–mRNA. This evidence provides valuable information for functional verification of the miRNAs and their targets in the metamorphosis of S. constricta.
References
Applebaum SL, Finn RN, Faulk CK, Holt GJ, Nunez BS (2012) Developmental expression, differential hormonal regulation and evolution of thyroid and glucocorticoid receptor variants in a marine acanthomorph teleost (Sciaenops ocellatus). Gen Comp Endocrinol 176:39–51
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233
Bogazzi F, Hudson LD, Nikodem VM (1994) A novel heterodimerization partner for thyroid hormone receptor. Peroxisome proliferator-activated receptor. J Biol Chem 269:11683–11686
Brown CL, Urbinati EC, Zhang W, Brown SB, Mccomb-Kobza M (2014) Maternal thyroid and glucocorticoid hormone interactions in larval fish development, and their applications in aquaculture. Rev Fish Sci Aquac 22:207–220
Campbell AH, Meritt DW, Franklin RB, Boone EL, Nicely CT, Brown BL (2011) Effects of age and composition of field-produced biofilms on oyster larval setting. Biofouling 27:255–265
Clare AS, Fusetani N, Jones MB (1998) Settlement and metamorphosis of marine invertebrate larvae - introduction. Biofouling 12:1–2
Conaco C, Neveu P, Zhou H, Arcila ML, Degnan SM, Degnan BM, Kosik KS (2012) Transcriptome profiling of the demosponge Amphimedon queenslandica reveals genome-wide events that accompany major life cycle transitions. BMC Genomics 13:209
Elias T, Andreas H (2018) Thyroid hormones accelerate initiation of skeletogenesis via MAPK (ERK1/2) in larval sea urchins (Strongylocentrotus purpuratus). Front Endocrinol 9:439
Friedlaender MR, Mackowiak SD, Li N, Chen W, Rajewsky N (2012) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40:37–52
Garcia-Lavandeira M, Silva A, Abad M, Pazos AJ, Sanchez JL, Perez-Paralle ML (2005) Effects of GABA and epinephrine on the settlement and metamorphosis of the larvae of four species of bivalve molluscs. J Exp Mar Biol Ecol 316:149–156
Geo P, Huang X, Feng L, Valentin A, Razvan S, Svetlana K, Yuandan L, Joseph W, Foo C, Babak PJB (2003) TIGR gene indices clustering tools (TGICL): a software\nsystem for fast clustering of large EST datasets. Bioinformatics 19:651–652
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Biotechnology OJN (2013) Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol 29:644
Henry JJ, Collin R, Perry KJ (2010) The slipper snail, Crepidula: an emerging lophotrochozoan model system. Biol Bull 218:211–229
Hettinger A, Sanford E, Hill TM, Russell AD, Sato KNS, Hoey J, Forsch M, Page HN, Gaylord B (2012) Persistent carry-over effects of planktonic exposure to ocean acidification in the Olympia oyster. Ecology 93:2758–2768
Huan P, Wang H, Liu B (2012) Transcriptomic analysis of the clam Meretrix meretrix on different larval stages. Mar Biotechnol 14:69–78
John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS (2004) Human microRNA targets. PLoS Biol 2:e363
Kalia DJC (2013) Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p)ppGpp signaling in bacteria and implications in pathogenesis. Chem Soc Rev 42:305–341
Kang XL, Zhang JY, Wang D, Zhao YM, Han XL, Wang JX, Zhao XF (2019) The steroid hormone 20-hydroxyecdysone binds to dopamine receptor to repress lepidopteran insect feeding and promote pupation. PLoS Genet 15:e1008331
Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10:126–139
Kozomara A, Birgaoanu M, Griffiths-Jones S (2019) miRBase: from microRNA sequences to function. Nucleic Acids Res 47:D155–D162
Li H, Bai L, Dong X, Qi X, Liu H, Yu D (2020a) SEM observation of early shell formation and expression of biomineralization-related genes during larval development in the pearl oyster Pinctada fucata. Comp Biochem Physiol D Genomics Proteomics 33
Li Z, Niu DH, Peng MX, Xiong Y, Ji J, Dong ZG, Li JL (2020b) Dopamine beta-hydroxylase and its role in regulating the growth and larval metamorphosis in Sinonovacula constricta. Gene 737:144418–144418
Liang X, Chen YR, Gao W, Guo XP, Ding DW, Yoshida A, Osatomi K, Yang JL (2018) Effects on larval metamorphosis in the mussel Mytilus coruscus of compounds that act on downstream effectors of G-protein-coupled receptors. J Mar Biol Assoc U K 98:333–339
Lukwambe B, Zhao L, Nicholaus R, Yang W, Zhu JY, Zheng ZM (2019) Bacterioplankton community in response to biological filters (clam, biofilm, and macrophytes) in an integrated aquaculture wastewater bioremediation system. Environ Pollut 254
Mccormick MI, Molony BW (1995) Influence of water temperature during the larval stage on size, age and body condition of a tropical reef fish at settlement. Mar Ecol Prog Ser 118:59–68
Niu DH, Wang F, Xie SM, Sun FY, Wang Z, Peng MX, Li JL (2016) Developmental transcriptome analysis and identification of genes involved in larval metamorphosis of the razor clam, Sinonovacula constricta. Mar Biotechnol 18:168–175
O’connor S, Moltschaniwskyj N, Bolch CJS, O’Connor W (2015) Assessment of temperature or salinity effects on larval development by catecholamine-induced metamorphosis of hatchery-reared flat oyster, Ostrea angasi (Sowerby 1871) larvae. Aquac Res 46:2501–2511
Okazaki Y, Shizuri Y (2000) Effect of inducers and inhibitors on the expression of bcs genes involved in cypris larval attachment and metamorphosis of the barnacles Balanus amphitrite. Int J Dev Biol 44:451–456
Parichy DM, Turner JM, Parker NB (2003) Essential role for puma in development of postembryonic neural crest-derived cell lineages in zebrafish. Dev Biol 256:221–241
Peng LH, Liang X, Xu JK, Dobretsov S, Yang JL (2020) Monospecific biofilms of Pseudoalteromonas promote larval settlement and metamorphosis of Mytilus coruscus. Sci Rep 10:2577–2577
Qi QZ, Yang MY (1984) The growth and development of the plankonic larvae, spats and young shells of Sinonovacula constricta. Taiwan Strait 000:92–101
Rey F, Silva Neto GM, Bueno-Pardo J, Bispo R, Calado R (2019) Supply and larval traits at metamorphosis of a coastal marine invertebrate with a bi-phasic life cycle under contrasting oceanographic conditions. Prog Oceanogr 178:102201
Romero MR, Phuong MA, Bishop C, Krug PJ (2012) Nitric oxide signaling differentially affects habitat choice by two larval morphs of the sea slug Alderia willowi: mechanistic insight into evolutionary transitions in dispersal strategies. J Exp Biol 216:1114–1125
Sanchez-Lazo C, Martinez-Pita I, Young T, Alfaro AC (2012) Induction of settlement in larvae of the mussel Mytilus galloprovincialis using neuroactive compounds. Aquaculture 344:210–215
Satuito CG, Natoyama K, Yamazaki M, Shimizu K, Fusetani N (1999) Induction of metamorphosis in the pediveliger larvae of the mussel Mytilus galloprovincialis by neuroactive compounds. Fish Sci 65:384–389
Scholz S, Renner P, Belanger SE, Busquet F, Toxicology MREJCRI (2012) Alternatives to in vivo tests to detect endocrine disrupting chemicals (EDCs) in fish and amphibians-screening for estrogen, androgen and thyroid hormone disruption. Crit Rev Toxicol 43:45–72
Schoonjans K, Staels B, Auwerx JJBEBA (1996) The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta 1302:93–109
Shikuma NJ, Antoshechkin I, Medeiros JM, Pilhofer M, Newman DK (2016) Stepwise metamorphosis of the tubeworm Hydroides elegans is mediated by a bacterial inducer and MAPK signaling. Proc Natl Acad Sci U S A 113:10097–10102
Su Y, Shi YF, Stolow MA, Shi YB (1998) Thyroid hormone induces apoptosis in primary cell cultures of tadpole intestine: cell type specificity and effects of extracellular matrix. J Cell Biol 139(6):1533–1543
Sun J, Wang SM, Li C, Ren YJ, Wang JQ (2014) Novel expression profiles of microRNAs suggest that specific miRNAs regulate gene expression for the sexual maturation of female Schistosoma japonicum after pairing. Parasit Vectors 7:177
Sun XJ, Liu ZH, Zhou LQ, Wu B, Yang AG, Tian JT (2020) Developmental dynamics of the larval muscle system of bay scallop (Argopecten irradians). J Ocean Univ China 19:386–392
Tremblay R, Christophersen G, Nadalini JB, Redjah I, Magnesen T, Andersen S (2020) Improving scallop (Pecten maximus and Placopecten magellanicus) spat production by initial larvae size and hydrodynamic cues used in nursery system. Aquaculture:516. https://doi.org/10.1016/j.aquaculture.2019.734650
Vogeler S, Wikfors GH, Li X, Veilleux D, Miller-Ezzy P, Joyce A (2019) Larval metamorphosis in four bivalve species in response to NMDA receptor ligands: the NMDA receptor pathway as potential regulator of bivalve transition to spat. Aquaculture 511:634173
Wang L, Shao YY, Ballock RT (2011) Leptin synergizes with thyroid hormone signaling in promoting growth plate chondrocyte proliferation and terminal differentiation in vitro. Bone 48:1022–1027
Weaver DM, Coghlan SM Jr, Zydlewski J (2018) The influence of nutrients from carcasses of sea lamprey (Petromyzon marinus) on larval growth and spawner abundance. Fish Bull 116:142–152
Xu F, Wang XT, Feng Y, Huang W, Wang W, Li L, Fang XD, Que HY, Zhang GF (2014) Identification of conserved and novel microRNAs in the pacific oyster Crassostrea gigas by deep sequencing. PLoS One 9
Yang JL, Li SH, Li YF, Liu ZW, Liang X, Bao WY, Li JL (2013) Effects of neuroactive compounds, ions and organic solvents on larval metamorphosis of the mussel Mytilus coruscus. Aquaculture 396:106–112
Yang BY, Pu F, Qin J, You WW, Ke CH (2014) Characterization of receptor of activated C kinase 1 (RACK1) and functional analysis during larval metamorphosis of the oyster Crassostrea angulata. Gene 537:294–301
Yang XX, Wong YH, Zhang Y, Zhang G, Qian PYJB (2018) Exploring the regulatory role of nitric oxide (NO) and the NO-p38MAPK/cGMP pathway in larval settlement of the bryozoan Bugula neritina. Biofouling 34:545–556
Yu CY, Kuo HCJJOBS (2019) The emerging roles and functions of circular RNAs and their generation. J Biomed Sci 26:29
Zhang Y, He LS, Zhang G, Xu Y, Lee OO, Matsumura K, Qian PY (2012) The regulatory role of the NO/cGMP signal transduction cascade during larval attachment and metamorphosis of the barnacle Balanus (=Amphibalanus) amphitrite. J Exp Biol 215:3813–3822
Zheng Z, Hao RJ, Xiong XW, Jiao Y, Deng YW, Du XD (2019) Developmental characteristics of pearl oyster Pinctada fucata martensii: insight into key molecular events related to shell formation, settlement and metamorphosis. BMC Genomics 20:122
Funding
This work was supported by the National Key R & D Plan “Blue Granary Science and Technology Innovation” special project (2019YFD0900700), Jiangsu Key Laboratory of Marine Biotechnology (HS2019002), and the National Natural Science Foundation of China (31472278).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 69 kb)
Rights and permissions
About this article

Cite this article
Niu, D., Li, B., Xie, S. et al. Integrated mRNA and Small RNA Sequencing Reveals Regulatory Expression of Larval Metamorphosis of the Razor Clam. Mar Biotechnol 22, 696–705 (2020). https://doi.org/10.1007/s10126-020-09993-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-020-09993-0