
Abstract
Long-chain (≥ C20) polyunsaturated fatty acids (LC-PUFA), such as eicosapentaenoic acid (20:5n-3, EPA) and docosahexaenoic acid (22:6n-3, DHA), are necessary for human health and are obtained from marine fish-derived oils. Marine fish are LC-PUFA-rich animals; however, many of them require LC-PUFA for growth. Therefore, it is suggested that they do not have sufficient ability to biosynthesize LC-PUFA. To evaluate in vivo LC-PUFA synthetic activity in fish cells, fish-derived cell lines from red sea bream (Pagrus major, PMS and PMF), Japanese flounder (Paralichthys olivaceus, HINAE), and zebrafish (Danio rerio, BRF41) were incubated with n-3 fatty acids labeled by radioisotopes or stable isotopes, and then, n-3 PUFA were analyzed by thin-layer chromatography or liquid chromatography-mass spectrometry. Labeled EPA and DHA were biosynthesized from labeled α-linolenic acid (18:3n-3) in BRF41, whereas they were not detected in PMS, PMF, or HINAE cells. We next cloned the fatty acid desaturase 2 (Fads2) cDNAs from PMF cells and zebrafish, expressed in budding yeasts, and then analyzed the substrate specificities of enzymes. As a result, we found that Fads2 from PMF cells was a ∆6/∆8 desaturase. Collectively, our study indicates that cell lines from red sea bream and Japanese flounder were not able to synthesize EPA or DHA by themselves, possibly due to the lack of ∆5 desaturase activity. Furthermore, this study provides a sensitive and reproducible non-radioactive method for evaluating LC-PUFA synthesis in fish cells using a stable isotope and liquid chromatography-mass spectrometry.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Vertebrates cannot de novo synthesize polyunsaturated fatty acids (PUFA); therefore, they must obtain PUFA from the diet. Docosahexaenoic acid (DHA, 22:6n-3) and eicosapentaenoic acid (EPA, 20:5n-3) are n-3 long-chain (≥ C20) PUFA (n-3 LC-PUFA) that have a double bond at the third carbon numbering from the methyl terminus. n-3 PUFA are important for the maintenance of human health, neural development, and treatment of cardiovascular diseases (Swanson et al. 2012; Calder 2014). DHA and EPA are used as nutritional supplements, and EPA is also used as a medicine for hyperlipidemia and arteriosclerosis obliterans in Japan. Marine fish are the main n-3 LC-PUFA sources because they accumulate n-3 LC-PUFA through marine food webs from lower trophic organisms (Khozin-Goldberg et al. 2011; Shulse and Allen 2011; Kabeya et al. 2018). EPA and DHA are abundant in marine phytoplankton and zooplankton, and so marine fish obtain n-3 PUFA from their diet (Hamilton et al. 2020). Marine fish also require LC-PUFA as essential fatty acids (EFA), although freshwater and diadromous fish require C18 PUFA as EFA (Tocher 2010). Specifically, red sea bream, Pagrus major, and Japanese flounder, Paralichthys olivaceus, both being high economic-value fish in Japan, require high levels of DHA and EPA for their normal growth (Takeuchi et al. 1990; Furuita et al. 1999).
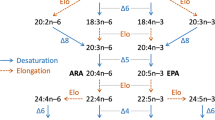
Vertebrates synthesize EPA and DHA using two types of responsible enzymes: fatty acid desaturase (Fads) and elongation of very long-chain fatty acids (Elovl) protein (Castro et al. 2016). The putative PUFA synthetic pathway of vertebrates is shown in Fig. 1. Fads enzymes introduce a double bond between the preexisting double bond and carboxyl terminus; thus, Fads are defined as “front-end” desaturases. Fatty acid desaturases that introduce the double bond between the preexisting bond and methyl terminus are called “methyl-end” desaturases. The function of Elovl is to add two carbons to the fatty acyl substrate through a condensing reaction. In vertebrates, three Elovl proteins, Elovl2, Elovl4, and Elovl5, are involved in LC-PUFA synthesis and show overlapping substrate specificities. Elovl2 and Elovl5 mainly participate in the elongation of C20–C22 and C18–C20 LC-PUFA to form C22–C24 and C20–C22 LC-PUFA, respectively. On the other hand, Elovl4 mainly elongates LC-PUFA, which has a chain length of more than 24. Methyl-end desaturases such as ω3 and ω6 desaturases are widely found in microorganisms, plants, and invertebrates (Pereira et al. 2003; Dar et al. 2017; Kabeya et al. 2018; Garrido et al. 2019b); thus, they are able to de novo synthesize n-3 or n-6 PUFA. Mammals have two major Fads involved in LC-PUFA biosynthesis: Fads1 and Fads2. Fads1 is responsible for ∆5 desaturase activity that synthesizes EPA and arachidonic acid (ARA, 20:4n-6) from 20:4n-3 and 20:3n-6, respectively. Fads2 possesses ∆6 desaturase activity that catalyzes the desaturation toward 18:3n-3 and 18:2n-6. LC-PUFA synthesis in vertebrates is initiated by ∆6 desaturation of 18:3n-3 and 18:2n-6 by Fads2 followed by an elongation reaction to form 20:4n-3 and 20:3n-6, and this pathway is termed the “∆6 pathway.” Since ∆8 desaturase activity of Fads2 was found in mammals (Park et al. 2009; Stroud et al. 2009) and fishes (Monroig et al. 2011), an alternative “∆8 pathway,” which is initiated by C18 elongation toward 18:3n-3 and 18:2n-6 followed by ∆8 desaturation by Fads2, has also been accepted. Almost all mammals are devoid of ∆4 desaturases; however, baboon Fads2 was reported to show ∆4 desaturase activity in human cells (Park et al. 2015). In addition, Fads2 is also crucial to synthesize DHA and 22:5n-6 from EPA and 22:4n-6 via the “Sprecher pathway” (Sprecher et al. 1995). In the Sprecher pathway, EPA is converted to 24:5n-3 by two sequential elongation steps, continuing ∆6 desaturation by Fads2 to form 24:6n-3 and two-carbon shortening by peroxisomal β-oxidation to produce DHA (Fig. 1).

Putative PUFA synthetic pathway of vertebrates. Solid and dashed arrows show desaturation and elongation reactions in the pathway, respectively. Mammals have Fads1 as Δ5 desaturase and Fads2 as Δ6 desaturase. Some teleosts have Fads2 with Δ5 or Δ4 desaturase activity. Vertebrates whose Fads2 desaturate C24 PUFA are able to synthesize DHA from 24:5n-3 followed by Δ6 desaturation and peroxisomal β-oxidation
In contrast to mammals, most Fads proteins isolated from teleost fishes have been phylogenetically classified into a Fads2 family. Since the fads1 gene was found in Elopomorpha and not in Clupeocephala or Osteoglossomorpha, this gene was likely lost after divergence to Teleostei (Lopes-Marques et al. 2018). To compensate for the loss of Fads1, some teleost Fads2 have developed additional desaturase activity. For example, bifunctional ∆6/∆5 desaturase activity was reported initially in zebrafish Danio rerio (Hastings et al. 2001), followed by multiple teleost species (Kuah et al. 2016; Janaranjani et al. 2018; Ferraz et al. 2019). Furthermore, Fads2 with ∆4 desaturase activity was first reported in rabbitfish Siganus canaliculatus (Li et al. 2010), and then in mainly freshwater fish (Fonseca-Madrigal et al. 2014; Oboh et al. 2017; Garrido et al. 2019a). These fish are able to convert DHA from EPA without synthesizing the intermediates 24:5n-3 and 24:6n-3. Notably, Fads2 with trifunctional ∆6/∆5/∆4 desaturase has been reported in freshwater flatfish Trinectes maculatus, Apionichthys finis, and Hypoclinemus mentalis (Matsushita et al. 2020). This diversity of Fads2 function may be dependent on habitat and trophic ecology; for example, S. canaliculatus is a marine herbivore, and therefore, it shows intravital LC-PUFA synthesis from C18 PUFA. Specifically, due to the poor supply of n-3 LC-PUFA such as EPA and DHA in lakes and rivers, Fads2 with ∆5 or ∆4 desaturase activity are mainly found in freshwater fish.
The yeast expression system has been used to examine in vitro enzymatic activity of Fads2, but this technique cannot be used to assess the intravital ability of LC-PUFA synthesis in fish. Radioisotope-labeled C18 PUFA have been used to analyze internal LC-PUFA synthetic activities in fish cell lines and primary cultured cells, suggesting weak ∆5 desaturase activity in a cell line of gilthead sea bream (Tocher and Ghioni 1999), low elongation activity toward C18 PUFA in a turbot cell line (Ghioni et al. 1999), and complete DHA synthetic ability in the brain and hepatic cells from freshwater flatfish (Matsushita et al. 2020). Although the method using radioisotope-labeling fatty acids as a precursor is useful for determining the internal LC-PUFA synthetic activity in fish cells, the use of radioisotopes has problems such as insufficient quantification, low resolution, and strict regulation.
In the present study, we investigated the n-3 LC-PUFA synthetic activity of fish at a cellular level using a conventional radioisotope method and new method using stable isotope-labeled fatty acids with liquid chromatography-electrospray ionization mass spectrometry (LC-ESI MS). Our data demonstrate that fish cell lines from red sea bream and Japanese flounder are not able to synthesize EPA or DHA from C18 PUFA, possibly due to the lack of ∆5 desaturase activity.
Materials and Methods
Cell lines
The red sea bream (Pagrus major) cell lines, PMS and PMF, developed from scale and fin tissue, respectively, were kindly provided by Dr. Jun Kurita (National Research Institute of Aquaculture, Fisheries Research Agency). The Japanese flounder (Paralichthys olivaceus) cell line, HINAE (RRID: CVCL_R908), developed from an embryo, was kindly provided by Dr. Hisae Kasai (Faculty of Fisheries Sciences, Hokkaido University) (Kasai and Yoshimizu 2001). The zebrafish (Danio rerio) cell line, BRF41 (RRID: CVCL_4131), developed from fin tissue was provided by the RIKEN Cell Bank (RIKEN Bioresource Center). Saccharomyces cerevisiae INVSc1 was purchased from Thermo Fisher Scientific.
Mediums
The medium for culturing fish cells was a bottle of Leibovitz’s L-15 medium (HyClone) (Leibovitz 1963), containing 10% BenchMark Fetal Bovine Serum (Gemini Bio) and 60 µg/mL kanamycin. The medium for culturing Escherichia coli was LB medium, and 50 µg/mL ampicillin was used for selection. The yeast culture medium YPD contained 2% (w/v) d-glucose, 2% (w/v) peptone, and 1% (w/v) yeast extract. URA(-)Glc (URA(-)Gal) medium was composed of 2% (w/v) d-glucose (d-galactose), 0.67% (w/v) yeast nitrogen without amino acids, 0.059% (w/v) CSM-ADE-HIS-LEU-TRP-URA, 0.002% (w/v) histidine, 0.01% (w/v) leucine, 0.002% (w/v) tryptophan, and 0.001% (w/v) adenine. Each medium plate was contained 1.5% (w/v) agar.
Cell Culture
Each fibroblast-like cell line was routinely maintained in Leibovitz’s L-15 medium supplemented with 2.05 mM l-glutamine, 60 µg/mL kanamycin as an antibiotic, and 10% fetal bovine serum (FBS). Cells were cultured in a 75 cm2 Nunc EasYFlask (Thermo Fischer Scientific) at 20 °C for PMS, PMF, and HINAE, and 33 °C for BRF41 cells.
Tracing the Metabolism of [1-14C]-Labeled 18:3n-3
BRF41 and PMS cells were seeded at a density of 1.1 × 106 cells into each 60-mm dish supplemented with 3 mL of the medium. After cells were attached to the bottom of the dish, 0.5 µCi of [1-14C]18:3n-3 was added to the dish. Cells were cultured for 2 days, and the medium was removed. Cells were washed twice with 1 mL of phosphate buffered saline (PBS) and then harvested. The fatty acids in the cells were extracted as fatty acid methyl esters (FAME). FAME were prepared by incubating the cells with 50 µL of toluene, 375 µL of methanol, and 0.3 mL of 8% methanolic-HCl for 90 min at 80 °C. FAME were extracted by the addition of 1 mL of hexane and 1 mL of distilled water. The mixture was centrifuged at 2000 rpm for 3 min, and the upper layer was collected. The lower layer was supplemented with 2 mL of hexane and centrifuged, and then, the upper phase was collected, promoting the recovery of FAME. The mixture was evaporated using the centrifugal concentrator VC-96 N (TAITEC). For TLC analyses, Silica gel 60 TLC plate (Merck) was sprayed with AgNO3 and activated at 110 °C for 30 min. FAME including 14C-labeled PUFA were added to the TLC plate and separated (solvent, acetonitrile:toluene, 97:3, v/v). The TLC plate was exposed to the imaging plate overnight at room temperature, and the radioactive signals were quantified using FLA-5000 (GE Healthcare, Japan).
Tracing the Metabolites of d5-Labeled PUFA
Cells (5 × 105) were sub-cultured in each well of the Nunc Cell-Culture Treated Multidishes 6-well plate (Thermo Fisher Scientific) containing 2 mL of culture medium. After being left for 24 h for cell colonization on the bottom, 1 µL of 1 µg/µL deuterium-labeled 18:3n-3 (d5-18:3n-3) or 20:5n-3 (d5-20:5n-3) was added to each well, while 1 µL of ethanol was used as a control. All experiments were performed in triplicate with three wells. After 48-h incubation at respective culture temperatures, the medium was aspirated, and cells were harvested. Cells were washed twice with Dulbecco’s PBS and stored at − 20 °C for further experiments.
Total Lipid Extraction and Alkaline Treatment
Cells were incubated at 37 °C for an hour with 400 µL of chloroform–methanol (2:1, v/v) and 10 µL of 5 µg/mL d8-arachidonic acid (internal standard). After centrifugation at 10,000 × g for 5 min, supernatants were collected in new tubes. Pellets were suspended with 400 µL of chloroform–methanol (1:1, v/v) followed by incubation and centrifugation. Supernatants from one sample were mixed and fully evaporated by VC-96 N. To convert lipids into fatty acids, 500 µL of 1.75 M methanolic-KOH was added to a sample and incubated at 65 °C for 2 h. After adding 4.5 mL of water and acidification with 6 M HCl to pH 4.0, the sample was applied to a Sep-Pak Vac RC (100 mg) C18 Cartridge (Waters), and the column was washed with 4 mL of water and 4 mL of hexane. A fatty acid fraction was collected by 2 mL of methyl formate, evaporated, and resuspended in 150 µL of methanol. After centrifugation at 10,000 × g for 10 min, the sample was transferred to a glass vial and analyzed by LC-ESI MS.
LC-ESI MS Analysis
LC-ESI MS was performed using a high-performance liquid chromatography (HPLC) system (Agilent Technologies) coupled with mass spectrometry (MS) apparatus (3200 QTRAP, Sciex). A binary solvent gradient with a flow rate of 200 µL/min was used to separate fatty acids by reverse-phase chromatography using XBridge BEH C18 (2.1 × 150 mm, 2.5 µm, Waters). The gradient was started with 60% buffer B (acetonitrile:2-propanol, 9:1, v/v) in buffer A (0.1% acetic acid) and was maintained for 3 min. The gradient reached 85% for 30 s, then 95% for 2 min, and was maintained for 6 min. The gradient was returned to the starting conditions for 30 s, and the column was equilibrated for 10 min before the next run. The fatty acids were quantified by multiple reaction monitoring (MRM) and calibration curves of known standards. The MRM pairs used for the experiments are shown in Supplementary Table 1.
Molecular Cloning of Red Sea Bream and Zebrafish Fads2
Total RNA was extracted from PMF cells using Sepasol RNA I Super G (nacalai tesque) and ReliaPrep RNA Cell Miniprep System (Promega). First-strand complementary DNA (cDNA) was synthesized from 2 µg of total RNA using PrimeScript II 1st strand cDNA Synthesis Kit (Takara Bio). The cDNA from PMF cells and adult zebrafish were used as templates together with primers to obtain open reading frames (ORF) of the fads2 gene. Polymerase chain reaction (PCR) was carried out using Tks Gflex DNA Polymerase (Takara Bio) under the following conditions: initial denaturation at 94 °C for 1 min with primers (Supplementary Table 2), followed by 35 cycles of denaturation of 98 °C for 10 s, annealing at 60 °C for 15 s, and extension at 68 °C for 2 min. Primers (HN31-F and HN31-R) designed on 5′ and 3′ outer regions of red sea bream fads2 (pmfads2) using a genome database of red sea bream (GenBank accession number: BDUH01000001–BDUH01886260). Zebrafish fads2 (drfads2) gene was cloned from the cDNA library using primers (HN45-F and HN46-R) following secondary PCR with primers (HN43-F and HN44-R) for conjugation with the pUC19 vector.
Sequence and Phylogenetic Analysis
The fads2 ORF sequences containing an overlapping region of the adduct of the linearized pUC19 vector were ligated with pUC19 vector using In-Fusion HD Cloning Kit (Takara Bio). The plasmids containing each fads2 gene were amplified in Escherichia coli HST08, purified using FastGene Plasmid Mini Kit (NIPPON Genetics), and then sequenced. The putative amino acid sequence of the red sea bream Fads2 protein was aligned with multiple functionally characterized Fads2 proteins using the method of MUSCLE (Edgar 2004). Phylogenetic analysis was performed using MEGAX software through the maximum-likelihood estimation algorithm with the Jones-Taylor-Thornton (JTT) substitution model (Jones et al. 1992; Kumar et al. 2018). The branch supporting values (%) were calculated from 1000 bootstrap replicates.
Functional Characterization of Fads2 from Red Sea Bream and Zebrafish
The fads2 ORF sequences in the pUC19 plasmid were amplified with primers (HN35-F and HN37-R for pmfads2; HN38-F and HN40-R for drfads2) as an overlapping region of adduct of the linearized pYES2CT yeast expression vector and then ligated with the pYES2CT vector by the abovementioned in-fusion reaction. The pYES2CT-fads2 plasmids were transformed into the budding yeast S. cerevisiae competent cells InvSc1 using the lithium acetate method (Schiestl and Gietz 1989). Briefly, InvSc1 cells were cultured at OD600 of 0.4 with 3 mL of YPD medium for 4 h and then recovered cells were incubated with 100 µL of OSB buffer containing 50% (w/v) polyethylene glycol, 0.2 M lithium acetate, 0.1 M dithiothreitol, 5 µL of 10 mg/mL salmon sperm DNA (Wako), and 0.5 µg of each plasmid. The mixture was incubated at 42 °C for an hour. Transformants were selected on URA(-)Glc plates and cultivated at 30 °C in 3 mL of URA(-)Glc medium. Precultured yeasts in URA(-)Gal medium in the logarithmic growth phase were diluted to OD600 of 0.4 and cultivated in 3 mL of URA(-)Gal medium with 0.1% TERGITOL type NP-40 (Sigma-Aldrich) and each fatty acid precursor, 18:3n-3, 18:2n-6, 20:3n-3, 20:2n-6, 20:4n-3, 20:3n-6, 22:5n-3, or 22:4n-6, at final a concentration of 0.5 mM. After 24-h culture at 30 °C, yeasts were harvested, washed twice with PBS, and lyophilized for further analysis.
Fatty Acid Analysis of Yeast
FAME were prepared by incubating the dried yeasts with 0.2 mL of toluene, 1.5 mL of methanol, and 0.3 mL of 8% methanolic-HCl at 45 °C overnight. After incubation, 1 mL of hexane and 1 mL of water were added; then, the mixture was centrifuged at 2000 rpm for 3 min, and the upper layer was collected. The lower layer was supplemented with 2 mL of hexane and centrifuged; the upper phase was collected and mixed with the former. FAME were separated and quantified using a GC-2014 gas chromatograph (Shimadzu) equipped with a 30-m × 25-mmID × 25-µm Rxi-5 ms column (Shinwa Chemical Industries) and a flame ionization detector. Nitrogen was used as a carrier gas with the thermal gradient of the oven from 160 to 220 °C at 2 °C/min. Individual FAME were identified by comparison with known standards. The desaturation conversion efficiencies from exogenously added fatty acid substrates were calculated by the proportion of substrate fatty acid converted to desaturated product as (product area/(product area + substrate area)) × 100.
Results
Analysis of LC-PUFA Synthesis in Fish Cells using [1-14C]18:3n-3 as a Precursor
We first evaluated the n-3 PUFA metabolism of red sea bream-derived PMS and zebrafish-derived BRF41 cells using 14C-labeled α-linolenic acid ([1-14C]18:3n-3). As shown in Fig. 2, [1-14C]18:3n-3 was metabolized to different n-3 PUFAs in each cell. 14C-labeled n-3 PUFAs corresponding to 18:4n-3, 20:4n-3, 20:5n-3 (EPA), 22:5n-3, and 22:6n-3 (DHA) were detected in BRF41 cells, indicating that this cell line was able to synthesize DHA from 18:3n-3. On the other hand, 14C-labeled 20:5n-3 (EPA), 22:5n-3, and 22:6n-3 (DHA) were not detected in PMS cells, suggesting that ∆5 desaturase activity responsible for conversion from 20:4n-3 to 20:5n-3 is missing in the red sea bream-derived cells.

Metabolism of [1-14C]18:3n-3 in fish cells. A Radioisotope-labeled FAME were separated by AgNO3-TLC. The radioactivity of the corresponding bands was quantified using a FLA 5000 Bio-imaging analyzer. B The relative radioactivity level of each band is expressed as a ratio against [1-14C]18:3n-3. The results are expressed as means ± SD (n = 3)
Analysis of LC-PUFA Synthesis in Fish Cells using d5-PUFA as a Precursor
To investigate detailed PUFA metabolic pathways in fish cells, an LC-ESI MS system that can separate each PUFA was established. As shown in Supplementary Fig. 1, each PUFA was successfully separated as a single peak under the conditions established in this study. Since only 5–10% of fatty acid was detected as a collisionally dissociated ion in the ESI–MS system, the pseudo-MRM value (Q1 = Q3) was also used for the detection of each PUFA. Metabolic analyses with d5-18:3n-3 as a precursor showed that BRF41 cells converted d5-18:3n-3 to d5-22:6n-3 (Fig. 3A). Simultaneously, d5-24:5n-3 and d5-24:6n-3 were detected in the presence of d5-20:5n-3 (Fig. 3B). These results indicate that BRF41 cells synthesize DHA (22:6n-3) through the Sprecher pathway. On the other hand, PMF and HINAE cells converted d5-18:3n-3 to d5-20:4n-3 through d5-18:4n-3, although d5-20:5n-3, and subsequent n-3 LC-PUFA were not detected (Fig. 3A). When d5-20:5n-3 was added to PMF and HINAE cells as a precursor, d5-22:5n-3 was detected, but d5-22:6n-3, d5-24:5n-3, and d5-24:6n-3 were not (Fig. 3B). These results indicate that PMF and HINAE cells also lacked C22 elongase activity, which converts 22:5n-3 to 24:5n-3.

d5-n-3 PUFA metabolic analyses using LC-ESI MS. PMF, HINAE, and BRF41 cells were incubated with d5-18:3n-3 A or d5-20:5n-3 B, and fatty acids were analyzed by LC-ESI MS. Each labeled fatty acid was quantified, and the relative amount against d5-18:3n-3 or d5-20:5n-3 is expressed on the Y-axis. Data are expressed as means ± SD (n = 3)
Sequence and Phylogenetic Analysis of Pmfads2 and Drfads2
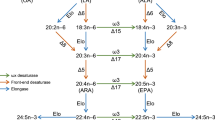
The full lengths of pmfads2 and drfads2 genes were obtained from cDNA of PMF cells and zebrafish, respectively. The deduced polypeptide sequence from 1338 bp ORF of pmfads2 (DDBJ Accession Number: LC589181.1) consisted of 445 amino acids containing typical conserved features of front-end desaturase (Fig. 4), namely, an N-terminal cytochrome b5 domain with the HPGG motif and three histidine boxes. Pmfads2 showed marked similarities to Fads2 from other Sparidae (Perciformes: Teleostei) species such as gilthead sea bream Sparus aurata (98%) and salema Sarpa salpa (98%), and moderate similarities to Fads2 from Japanese flounder (84%) and zebrafish (69%). The phylogenetic analysis comparing a variety of desaturases from teleosts showed that Pmfads2 was clustered closely with other Fads2 proteins from Sparidae species (100% bootstraps) including gilthead sea bream, salema, and black sea bream Acanthopagrus schlegelii with ∆6 desaturase activity (Fig. 5).

Multiple alignments of the polypeptide sequences of fish Fads2. The amino acid sequence of Pmfads2 was aligned with those of Δ6 desaturases from gilthead sea bream S. aurata (GenBank accession number: AAL17639.1), salema S. salpa (QAY29227.1), and Japanese flounder P. olivaceus (AJG36440.1), and a Δ6/Δ5 desaturase from zebrafish D. rerio (AAG25710.1). The amino acid residues of three histidine boxes (HXXXH, HXXHH, QXXHH) have a solid underline, and cytochrome b5 domain containing the HPGG motif has a dashed underline

Phylogenetic tree of characterized Fads2 from fish and mammals. Phylogenetic analysis was performed using MEGAX software through the maximum-likelihood method. The significance of each branch was tested by bootstrapping with 1000 replicates. The horizontal branch length is proportional to the amino acid substitution rate per region. The characterized function of each Fads2 is represented as the regioselectivity with “Dx” between the scientific name and GenBank accession number. The groups of Teleostei and Sparidae are indicated with marked nodes
Functional Characterization of Pmfads2 and Drfads2
We used the zebrafish fads2 (∆6/∆8/∆5 desaturase) gene as a positive control of the experiment (Hastings et al. 2001; Monroig et al. 2011). The substrate regioselectivity of the newly cloned Pmfads2 was determined by heterologous expression in yeast S. cerevisiae. Fatty acid profiles of the yeast transformed with pYES2CT mock vector were composed of endogenous fatty acids containing 16:0, 16:1n-7, 18:0, and 18:1n-9, and exogenously added PUFA (Supplementary Fig. 2). This limitation of PUFA metabolism in the yeast indicated the complete lack of enzymatic activity toward PUFA substrates used in this study. The substrate specificity of Drfads2 and Pmfads2 toward fatty acid substrates is shown in Table 1. Both Fads2s showed ∆6 desaturase activity toward 18:3n-3 and 18:2n-6, indicating that they were able to produce 18:4n-3 and 18:3n-6 in transformed yeast. Since some Fads2 were also reported to show ∆8 desaturase activity (Monroig et al. 2011; Kabeya et al. 2017), we also evaluated ∆8 desaturase activity using 20:3n-3 and 20:2n-6. As shown in Table 1, weak but significant ∆8 desaturase activity was detected in both drfads2 and pmfads2-expressing yeasts, indicating that Pmfads2 is a bifunctional ∆6/∆8 desaturase like other Fads2 in teleosts. ∆5 desaturase activity toward 20:4n-3 and 20:3n-6 to produce 20:5n-3 and 20:4n-6 was detected only in yeast expressing Drfads2, and not in yeast expressing Pmfads2. ∆4 desaturation toward 22:5n-3 and 22:4n-6 was not observed in either strain. As zebrafish fads2 showed ∆6/∆8/∆5 desaturase activity, we successfully reproduced the results reported by (Hastings et al. 2001) and (Monroig et al. 2011). Considering the fatty acid conversion rate, n-3 PUFA were more efficiently converted than n-6 PUFA in ∆6 (21.1% > 13.6%) and ∆5 (6.1% > 1.7%) desaturase activities of Drfads2 and ∆6 (20.2% > 10.6%) desaturase activity of Pmfads2.
Discussion
In the present study, we developed a method to evaluate endogenous LC-PUFA synthetic activity in fish cells using the LC-ESI MS system with deuterium-labeled fatty acids as a precursor and revealed that red sea bream and Japanese flounder could not synthesize EPA or DHA because of the loss of the ∆5 desaturase activity.
The experiments using radioisotope-labeled fatty acid [1-14C]18:3n-3 suggest the lack of ∆5 desaturase activity in red sea bream. Quantitative experiments with deuterium-labeled fatty acids suggest the inability of red sea bream and Japanese flounder to synthesize EPA and DHA. In the case of zebrafish, ∆6/∆8/∆5 desaturase was identified (Hastings et al. 2001; Monroig et al. 2011), and Elovl5 and Elovl2, which are responsible for elongation of C18–C20 and C20–C22 PUFA, respectively, were also identified (Agaba et al. 2004; Monroig et al. 2009). There are consistent with our observation that BRF41 cells convert 18:3n-3 to DHA. The intake of sufficient DHA is difficult for freshwater fish because of their short food chain length and poor supply from primary producers such as microalgae (Ishikawa et al. 2019); therefore, zebrafish can synthesize DHA from 18:3n-3. The results obtained from PMF and HINAE cells suggest that red sea bream and Japanese flounder are not able to synthesize EPA from 20:4n-3 or DHA from 22:5n-3, possibly due to lack of ∆5 desaturase and C22 elongase activity, respectively. Fads2 of Japanese flounder was reported as a ∆6/∆8 desaturase (Kabeya et al. 2017), and Fads2 of red sea bream was also revealed as a ∆6/∆8 desaturase in the present study. Thus, both cells stopped the conversion of 18:3n-3 at 20:4n-3.
In the case of aquaculture, Sarker et al. reported that culturing red sea bream on a special diet replacing two-thirds of fish oil with canola oil reduced the proportion of EPA and DHA in the whole body (Sarker et al. 2011), suggesting that red sea bream has no mechanism that compensates for EPA/DHA deprivation. In vertebrates, the elongation step from 22:5n-3 to 24:5n-3 is mainly carried out by Elovl2. However, in marine teleosts, Elovl2 has been found only in a limited number of marine fish belonging to salmonids and Otocephala and not found in Neoteleostei including Sparidae (Monroig et al. 2009; Machado et al. 2018; Datsomor et al. 2019; Ferraz et al. 2019). In addition, an in vitro study using trout hepatocytes demonstrated that conversion from EPA to DHA went through C24 PUFA (Buzzi et al. 1997). Elovl5 also could elongate C22 PUFA, although this contribution seems to be small compared with Elovl2. In particular, Elovl2, not Elovl5, is essential for DHA biosynthesis in zebrafish (Liu et al. 2020). Kabeya et al. suggested that Japanese flounder does not have Elovl2, and thus, it is not capable of synthesizing DHA (Kabeya et al. 2017). We searched for elovl2-like genes in the genome database of red sea bream (Sawayama et al. 2017), but no genes were found. On the other hand, vertebrates synthesize very long-chain (≥ C24) PUFA (VLC-PUFA) from LC-PUFA using Elovl4, and this enzyme has been reported not only in freshwater and diadromous fish but also marine fish (Monroig et al. 2010; Carmona-Antoñanzas et al. 2011; Jin et al. 2017). Elovl4 is able to elongate C22 PUFA; however, the activity is low compared with that toward VLC-PUFA (≥ C24). Furthermore, Elovl4 is expressed strongly in the eye and brain but weakly in the skin, and this suggests inadequate expression in the cell lines of PMF and HINAE from the fin or embryo to elongate 22:5n-3 to 24:5n-3. These facts suggest that many marine fish cannot synthesize DHA from 22:5n-3 because they lack the ability to convert 24:5n-3 from 22:5n-3 unless they have ∆4 desaturase.
The newly cloned full-length cDNA of pmfads2 encodes a protein of 445 amino acids containing typical features of front-end desaturase. An amino acid sequence of Pmfads2, which has ∆6 desaturase activity, is similar to other ∆6 desaturases from carnivorous Sparidae species. Pmfads2 is a ∆6/∆8 desaturase that has a greater affinity for n-3 substrates than n-6 substrates, and this tendency of greater substrate affinities has been reported in other marine fish Fads2 and mammal Fads2, suggesting that Fads2 has evolved to produce more n-3 LC-PUFA such as EPA and DHA (Brenner 1974; Zheng et al. 2009). Furthermore, Fads2 of the herbivorous Sparidae species salema is also a ∆6/∆8 desaturase (Garrido et al. 2019a), indicating that the regioselectivity of Fads2 is dependent on the phylogenetic distance rather than habitat environment. Sparidae species possessing ∆5 or ∆4 desaturases have not been classified to date. On the other hand, ∆6 desaturation is required for desaturation of not only 18:3n-3 but also 24:5n-3 to synthesize DHA via the Sprecher pathway. However, this ability is weak or absent in marine fish (Oboh et al. 2017), suggesting that this is one of the critical points for recurrent freshwater fish to settle and colonize freshwater habitats (Ishikawa et al. 2019).
Collectively, the present study shows detailed n-3 LC-PUFA synthetic activity of cultured cells of red sea bream, Japanese flounder, and zebrafish. The results obtained in this study indicate that the fish are not able to synthesize EPA and DHA from 18:3n-3, possibly due to the lack of ∆5 desaturase. It is well-known that LC-PUFA is mainly synthesized in the liver; when we analyze the LC-PUFA synthesis pathway using the established cell lines, we always need to evaluate the results very carefully with that in mind. The method reported in this study is simple and reliable, and we are able to apply it to analyze LC-PUFA synthesis not only in cell lines but also in living fish or organ culture. Furthermore, the present study advances our understanding of the link between heterologous expression of Fads2 in yeasts and internal LC-PUFA synthetic ability in fish cells. The simplified technology using cell lines, a stable isotope, and LC-ESI MS will facilitate the quantitative analysis of LC-PUFA synthesis in fish without genetic data and might contribute to the discovery of novel desaturases that exhibit no similarity with known desaturases.
Availability of Data and Material
All data not present in the manuscript is available upon request (correspondence to N.O., nokino@agr.kyushu-u.ac.jp).
References
Agaba M, Tocher DR, Dickson CA et al (2004) Zebrafish cDNA encoding multifunctional fatty acid elongase involved in production of eicosapentaenoic (20:5n–3) and docosahexaenoic (22:6n–3) acids. Mar Biotechnol 6:251–261
Brenner RR (1974) The oxidative desaturation of unsaturated fatty acids in animals. Mol Cell Biochem 3:41–52
Buzzi M, Henderson RJ, Sargent JR (1997) Biosynthesis of docosahexaenoic acid in trout hepatocytes proceeds via 24-carbon intermediates. Comp Biochem Physiol B Biochem Mol Biol 116:263–267
Calder PC (2014) Very long chain omega-3 (n-3) fatty acids and human health. Eur J Lipid Sci Technol 116:1280–1300
Carmona-Antoñanzas G, Monroig Ó, Dick JR et al (2011) Biosynthesis of very long-chain fatty acids (C>24) in Atlantic salmon: cloning, functional characterisation, and tissue distribution of an Elovl4 elongase. Comp Biochem Physiol B Biochem Mol Biol 159:122–129
Castro LFC, Tocher DR, Monroig O (2016) Long-chain polyunsaturated fatty acid biosynthesis in chordates: Insights into the evolution of Fads and Elovl gene repertoire. Prog Lipid Res 62:25–40
Dar AA, Choudhury AR, Kancharla PK, Arumugam N (2017) The FAD2 gene in plants: occurrence, regulation, and role. Front Plant Sci 8:1789
Datsomor AK, Zic N, Li K et al (2019) CRISPR/Cas9-mediated ablation of elovl2 in Atlantic salmon (Salmo salar L.) inhibits elongation of polyunsaturated fatty acids and induces Srebp-1 and target genes. Sci Rep 9:7533
Edgar RC (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Ferraz RB, Kabeya N, Lopes-Marques M et al (2019) A complete enzymatic capacity for long-chain polyunsaturated fatty acid biosynthesis is present in the Amazonian teleost tambaqui, Colossoma macropomum. Comp Biochem Physiol B Biochem Mol Biol 227:90–97
Fonseca-Madrigal J, Navarro JC, Hontoria F et al (2014) Diversification of substrate specificities in teleostei Fads2: characterization of ∆4 and ∆6∆5 desaturases of Chirostoma estor. J Lipid Res 55:1408–1419
Furuita H, Konishi K, Takeuchi T (1999) Effect of different levels of eicosapentaenoic acid and docosahexaenoic acid in Artemia nauplii on growth, survival and salinity tolerance of larvae of the Japanese flounder, Paralichthys olivaceus. Aquaculture 170:59–69
Garrido D, Kabeya N, Betancor MB et al (2019a) Functional diversification of teleost Fads2 fatty acyl desaturases occurs independently of the trophic level. Sci Rep 9:11199
Garrido D, Kabeya N, Hontoria F et al (2019b) Methyl-end desaturases with ∆12 and ω3 regioselectivities enable the de novo PUFA biosynthesis in the cephalopod Octopus vulgaris. Biochim Biophys Acta Mol Cell Biol Lipids 1864:1134–1144
Ghioni C, Tocher DR, Bell MV et al (1999) Low C18 to C20 fatty acid elongase activity and limited conversion of stearidonic acid, 18:4(n-3), to eicosapentaenoic acid, 20:5(n-3), in a cell line from the turbot, Scophthalmus maximus. Biochim Biophys Acta Mol Cell Biol Lipids 1437:170–181
Hamilton HA, Newton R, Auchterlonie NA, Müller DB (2020) Systems approach to quantify the global omega-3 fatty acid cycle. Nat Food 1:59–62
Hastings N, Agaba M, Tocher DR et al (2001) A vertebrate fatty acid desaturase with ∆5 and ∆6 activities. Proc Natl Acad Sci USA 98:14304–14309
Ishikawa A, Kabeya N, Ikeya K et al (2019) A key metabolic gene for recurrent freshwater colonization and radiation in fishes. Science 364:886–889
Janaranjani M, Mah MQ, Kuah MK et al (2018) Capacity for eicosapentaenoic acid and arachidonic acid biosynthesis in silver barb (Barbonymus gonionotus): functional characterisation of a Δ6/Δ8/Δ5 Fads2 desaturase and Elovl5 elongase. Aquaculture 497:469–486
Jin M, Monroig Ó, Navarro JC et al (2017) Molecular and functional characterisation of two elovl4 elongases involved in the biosynthesis of very long-chain (>C24) polyunsaturated fatty acids in black seabream Acanthopagrus schlegelii. Comp Biochem Physiol B Biochem Mol Biol 212:41–50
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8:275–282
Kabeya N, Chiba M, Haga Y et al (2017) Cloning and functional characterization of fads2 desaturase and elovl5 elongase from Japanese flounder Paralichthys olivaceus. Comp Biochem Physiol B Biochem Mol Biol 214:36–46
Kabeya N, Fonseca MM, Ferrier DEK et al (2018) Genes for de novo biosynthesis of omega-3 polyunsaturated fatty acids are widespread in animals. Sci Adv 4:1–9
Kasai H, Yoshimizu M (2001) Establishment of two Japanese fIounder embryo cell lines. Bull Fish Sci Hokkaido Univ 52:67–70
Khozin-Goldberg I, Iskandarov U, Cohen Z (2011) LC-PUFA from photosynthetic microalgae: occurrence, biosynthesis, and prospects in biotechnology. Appl Microbiol Biotechnol 91:905–915
Kuah MK, Jaya-Ram A, Shu-Chien AC (2016) A fatty acyl desaturase (fads2) with dual Δ6 and Δ5 activities from the freshwater carnivorous striped snakehead Channa striata. Comp Biochem Physiol A Mol Integr Physiol 201:146–155
Kumar S, Stecher G, Li M et al (2018) MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Leibovitz A (1963) The growth and maintenance of tissue-cell cultures in free gas exchange with the atmosphere. Am J Epidemiol 78:173–180
Li Y, Monroig O, Zhang L et al (2010) Vertebrate fatty acyl desaturase with ∆4 activity. Proc Natl Acad Sci USA 107:16840–1684
Liu C, Ye D, Wang H et al (2020) Elovl2 but not Elovl5 is essential for the biosynthesis of docosahexaenoic acid (DHA) in zebrafish: insight from a comparative gene knockout study. Mar Biotechnol 22:613–619
Lopes-Marques M, Kabeya N, Qian Y et al (2018) Retention of fatty acyl desaturase 1 (fads1) in Elopomorpha and Cyclostomata provides novel insights into the evolution of long-chain polyunsaturated fatty acid biosynthesis in vertebrates. BMC Evol Biol 18:157
Machado AM, Tørresen OK, Kabeya N et al (2018) “Out of the can”: a draft genome assembly, liver transcriptome, and nutrigenomics of the European Sardine, Sardina pilchardus. Genes (basel) 9:1–13
Matsushita Y, Miyoshi K, Kabeya N et al (2020) Flatfishes colonised freshwater environments by acquisition of various DHA biosynthetic pathways. Commun Biol 3:1–9
Monroig Ó, Li Y, Tocher DR (2011) Delta-8 desaturation activity varies among fatty acyl desaturases of teleost fish: High activity in delta-6 desaturases of marine species. Comp Biochem Physiol B Biochem Mol Biol 159:206–213
Monroig O, Rotllant J, Cerdá-Reverter JM et al (2010) Expression and role of Elovl4 elongases in biosynthesis of very long-chain fatty acids during zebrafish Danio rerio early embryonic development. Biochim Biophys Acta Mol Cell Biol Lipids 1801:1145–1154
Monroig Ó, Rotllant J, Sánchez E et al (2009) Expression of long-chain polyunsaturated fatty acid (LC-PUFA) biosynthesis genes during zebrafish Danio rerio early embryogenesis. Biochim Biophys Acta Mol Cell Biol Lipids 1791:1093–1101
Oboh A, Kabeya N, Carmona-Antoñanzas G et al (2017) Two alternative pathways for docosahexaenoic acid (DHA, 22:6n–3) biosynthesis are widespread among teleost fish. Sci Rep 7:3889
Park HG, Park WJ, Kothapalli KSD, Brenna JT (2015) The fatty acid desaturase 2 (FADS2) gene product catalyzes Δ4 desaturation to yield n-3 docosahexaenoic acid and n-6 docosapentaenoic acid in human cells. FASEB J 29:3911–3919
Park WJ, Kothapalli KSD, Lawrence P et al (2009) An alternate pathway to long-chain polyunsaturates: the FADS2 gene product Δ8-desaturates 20:2n–6 and 20:3n–3. J Lipid Res 50:1195–1202
Pereira SL, Leonard AE, Mukerji P (2003) Recent advances in the study of fatty acid desaturases from animals and lower eukaryotes. Prostaglandins Leukot Essent Fat Acids 68:97–106
Sarker MA-A, Yamamoto Y, Haga Y et al (2011) Influences of low salinity and dietary fatty acids on fatty acid composition and fatty acid desaturase and elongase expression in red sea bream Pagrus major. Fish Sci 77:385–396
Sawayama E, Tanizawa S, Kitamura S et al (2017) Identification of quantitative trait loci for resistance to RSIVD in red sea bream (Pagrus major). Mar Biotechnol 19:601–613
Schiestl RH, Gietz RD (1989) High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet 16:339–346
Shulse CN, Allen EE (2011) Widespread occurrence of secondary lipid biosynthesis potential in microbial lineages. PLoS ONE 6:e20146
Sprecher H, Luthria DL, Mohammed BS, Baykousheva SP (1995) Reevaluation of the pathways for the biosynthesis of polyunsaturated fatty acids. J Lipid Res 36:2471–2477
Stroud CK, Nara TY, Roqueta-Rivera M et al (2009) Disruption of FADS2 gene in mice impairs male reproduction and causes dermal and intestinal ulceration. J Lipid Res 50:1870–1880
Swanson D, Block R, Mousa SA (2012) Omega-3 fatty acids EPA and DHA : health benefits throughout life. Adv Nutr 3:1–7
Takeuchi T, Toyota M, Satoh S, Watanabe T (1990) Requirement of juvenile red seabream Pagrus major for eicosapentaenoic and docosahexaenoic acids. Nippon Suisan Gakkaishi 56:1263–1269
Tocher DR (2010) Fatty acid requirements in ontogeny of marine and freshwater fish. Aquac Res 41:717–732
Tocher DR, Ghioni C (1999) Fatty acid metabolism in marine fish: low activity of fatty acyl Δ5 desaturation in gilthead sea bream (Sparus aurata) cells. Lipids 34:433–440
Zheng X, Ding Z, Xu Y et al (2009) Physiological roles of fatty acyl desaturases and elongases in marine fish: Characterisation of cDNAs of fatty acyl Δ6 desaturase and elovl5 elongase of cobia (Rachycentron canadum). Aquaculture 290:122–131
Acknowledgements
We thank Dr. Jun Kurita (National Research Institute of Aquaculture, Fisheries Research and Education Agency) for providing us the PMS and PMF cell lines. We are also grateful to Dr. Hisae Kasai (Faculty of Fisheries Sciences, Hokkaido University) who provided us the HINAE cell line.
Funding
This work was supported in part by the Sasakawa Scientific Research Grant from The Japan Science Society (to H.Y.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nyunoya, H., Noda, T., Kawamoto, Y. et al. Lack of ∆5 Desaturase Activity Impairs EPA and DHA Synthesis in Fish Cells from Red Sea Bream and Japanese Flounder. Mar Biotechnol 23, 472–481 (2021). https://doi.org/10.1007/s10126-021-10040-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-021-10040-9