
Abstract
Possessing powerful adaptive capacity and a pleasant taste, spotted sea bass (Lateolabrax maculatus) has a broad natural distribution and is one of the most popular mariculture fish in China. However, the genetic improvement program for this fish is still in its infancy. Growth is the most economically important trait and is controlled by quantitative trait loci (QTL); thus, the identification of QTLs and genetic markers for growth-related traits is an essential step for the establishment of marker-assisted selection (MAS) breeding programs. In this study, we report the first high-density linkage map of spotted sea bass constructed by sequencing 333 F1 generation individuals in a full-sib family using 2b-RAD technology. A total of 6883 SNP markers were anchored onto 24 linkage groups, spanning 2189.96 cM with an average marker interval of 0.33 cM. Twenty-four growth-related QTLs, including 13 QTLs for body weight and 11 QTLs for body length, were successfully detected, with phenotypic variance explained (PVE) ranging from 5.1 to 8.6%. Thirty potential candidate growth-related genes surrounding the associated SNPs were involved in cell adhesion, cell proliferation, cytoskeleton reorganization, calcium channels, and neuromodulation. Notably, the fgfr4 gene was detected in the most significant QTL; this gene plays a pivotal role in myogenesis and bone growth. The results of this study may facilitate marker-assisted selection for breeding populations and establish the foundation for further genomic and genetic studies investigating spotted sea bass.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spotted sea bass (Lateolabrax maculatus), as a eurythermic and euryhaline fish species, is widely distributed along the Chinese coasts, extending south to borders between China and Vietnam and north to the southeastern coast of South Korea (Wang et al. 2016a). With high nutritional values and a pleasant taste (Chen et al. 2019b), L. maculatus has become one of the most popular mariculture fish in China, and the production of this fish has reached 156,000 tons a year (MOA 2018). However, the lack of selective breeding of spotted sea bass leads to the degeneration of genetic characteristics, such as the decline in the growth rate and decreased disease-resistant ability (Wang et al. 2017). In addition, the long-term generation interval (3–4 years) hindered the progress of traditional breeding in this fish species. Marker-assisted selection (MAS), the selection method based on DNA markers that are tightly linked to quantitative trait loci (QTL) for traits of interest, has been suggested to be an accurate and efficient way to improve traits that are difficult to select by traditional breeding programs (Zhu et al. 2019). A number of QTL studies on various economic traits, including growth (Wang et al. 2019a, c), sex determination (Wang et al. 2019b; Zhou et al. 2020), disease resistance (Kong et al. 2019; Wu et al. 2019; Zhang et al. 2020), stress tolerance (Li et al. 2017; Jiang et al. 2019), and body color (Li et al. 2019a), have been successfully conducted for aquaculture fish species; however, little work has been performed for spotted sea bass. In the last 2 years, large-scale genomic resources of spotted sea bass have become available, including whole-genome sequencing data (Shao et al. 2018; Chen et al. 2019a), transcriptome databases (Tian et al. 2019c; Shen et al. 2019; Zhou et al. 2019a; Cai et al. 2020), and studies of functional genes (Wang et al. 2018; Zhang et al. 2019a, b, d, e; Liu et al. 2019; Tian et al. 2019a, b; Li et al. 2019b; Fan et al. 2019a, b; Zhou et al. 2019b). However, the linkage map of spotted sea bass has not been fully developed, and this map is essential for the identification of QTLs of economically important traits (Feng et al. 2018) and further facilitates MAS breeding programs.
Growth is the most economically important trait affecting aquaculture fish and exerts a direct influence on production. Combining the high-density linkage map with related phenotypic data, several growth-related QTLs and genetic markers have been identified in many aquaculture species, such as Asian sea bass (Lates calcarifer) (Wang et al. 2015), common carp (Cyprinus carpio) (Peng et al. 2016), bighead carp (Hypophthalmichthys nobilis) (Fu et al. 2016), crucian carp (Carassius auratus) (Liu et al. 2017), mandarin fish (Siniperca chuatsi) (Sun et al. 2017), pikeperch (Sander lucioperca) (Guo et al. 2018), yellow drum (Nibea albiflora) (Qiu et al. 2018), largemouth bass (Micropterus salmoides) (Dong et al. 2019), snapper (Chrysophrys auratus) (Ashton et al. 2019), channel catfish (Ictalurus punctatus) (Zhang et al. 2019c), Pacific white shrimp (Litopenaeus vannamei) (Huang et al. 2020), and Takifugu (Takifugu bimaculatus) (Shi et al. 2020). In addition, with the availability of whole-genome annotation for many fish species, several candidate genes for growth-related traits have been characterized; for example, in L. calcarifer, six growth-related QTLs were detected, and ACOX1 was considered a vital candidate functional gene (Wang et al. 2015). In C. auratus, eight QTLs were identified for body weight, and five potential genes, including EGF-like domain, immunoglobulin-like, C2H2 zinc finger, TGF-beta, and protein kinase (ATP binding site), were detected in the candidate regions (Liu et al. 2017). A total of six candidate QTLs were presented in I. punctatus for growth-related traits, in which three growth-related genes were identified, including megf9, npffr1, and gas1 (Zhang et al. 2019c). For C. carpio, fourteen QTL regions were detected for body weight, while four QTLs were identified for body length. Important regulators, such as KISS2, IGF1, SMTLB, and NPFFR1, were regarded as candidate genes for both growth traits (Peng et al. 2016). The growth-related genetic markers and candidate genes generated based on QTL mapping provide a useful basis for fish MAS breeding programs. However, further genetic studies of economically important traits of spotted sea bass lag behind.
Accordingly, the objectives for this study are (1) constructing the first high-density linkage map for L. maculatus, (2) performing fine QTL mapping for growth-related traits, including body weight and body length, and (3) identifying a list of candidate SNP markers and genes associated with growth-related traits. Our results provide a valuable resource for elucidating the genetic mechanisms of growth and accelerating the genetic breeding of L. maculatus.
Materials and Methods
Ethics Statement
All experimental procedures involving the fish were conducted in accordance with approved guidelines of the respective Animal Research and Ethics Committees of Ocean University of China (Permit Number: 20141201). The present study did not include endangered or protected species.
Resource Family and DNA Extractions
L. maculatus adults were collected from Qingdao, Shandong, China, and considered candidate broodstock for establishing mapping families. A total of 7 candidate full-sib families were produced in November 2016 through mating 7 sires and 7 dams by artificial fertilization. F1 larval fish of those families were raised at Shuangying Aquatic Seedling Co., Ltd., Lijin, Shandong, China. After 1 year of cultivation, one family exhibiting high within-family phenotypic variation in growth was selected for linkage map and QTL analyses. In total, 333 1-year-old progenies were chosen for linkage map construction. The body weights and body lengths of these fish were measured and recorded individually for subsequent growth-related QTL analyses (Supplementary Table S1). A piece of pectoral fin of parents and progenies was separately sampled and stored in 95% ethanol for DNA extractions. Following a traditional phenol-chloroform protocol (Zhan et al. 2007), genomic DNA of each sample was obtained using the CTAB method and purified with RNA digestion (TaKaRa). The concentration and quality of genomic DNA were determined by a Biodropsis BD-1000 nucleic acid analyzer (OSTC, Beijing) and electrophoresis in 1% agarose gel. Finally, high-quality gDNA was prepared for further sequencing.
2b-RAD Library Construction and Sequencing
2b-RAD libraries were prepared for two parents and 333 progenies following the protocol (Wang et al. 2016b). Briefly, the genomic DNA from each individual was first digested with BsaXI (New England Biolabs); then, the digestion products were ligated to adaptors, and the ligated fragments were amplified via polymerase chain reaction (PCR). Finally, the purified amplification products were further digested to generate cohesive ends and then ligated in a predefined order to produce five concatenated tags for paired-end sequencing. Pooled sequencing was carried out on an Illumina HiSeq Xten platform at Qingdao Oebiotech Co. Ltd.
Data Filtering and Genotyping
After sequencing, these raw paired-end reads were first filtered to remove reads that had N bases (ambiguous bases) greater than 8% of the total bases, and reads over 15% of the length were less than Q30. The paired-end reads were merged by Pear software (Version 0.9.6). The merged reads were processed using a custom Perl script to trim adaptor sequences and the terminal 3-bp bases. The remaining five-tag reads were divided into single-tag reads using a Perl script and then assigned to each individual via tag position and length. Finally, clean reads with a length of 27 bp were used for genotyping using the RADtyping software package (Fu et al. 2013). Raw SNP data were filtered as follows. (1) The SNP sites with one or four base types were removed. (2) The sites with minor allele frequency < 0.05 were discarded. (3) The sites with less than 80% of progeny samples being genotyped were filtered. (4) The tags containing more than one SNP were discarded. (5) SNPs with nn × np (heterozygous in female parent), lm × ll (heterozygous in male parent, and hk × hk (heterozygous in both parents) segregation patterns were retained. The SNP annotations were performed using SnpEff software (version 4.1) (Cingolani et al. 2012) with the genome file of L. maculatus (ASM402866v1).
Linkage Map Construction
The Mendelian segregation pattern of each SNP was examined using χ2 tests. Both female and male linkage maps were constructed under a logarithm of odds (LOD) threshold of 5.0 using JoinMap 4.1 (Van Ooijen 2006). Recombination rates were calculated by the regression mapping algorithm and were converted into map distances in centimorgans (cM) using the Kosambi mapping function. The sex-averaged map was produced by integrating the two sex-specific maps using MergeMap software (http://www.mergemap.org/). All genetic linkage maps were drawn via MapChart v 2.3 software (Voorrips 2002).
QTL Analyses
Combining phenotype data with the high-density linkage map, QTL mapping for growth-related traits was performed with MapQTL version 6.0 (Van Ooijen and Kyazma 2009). The LOD scores of each LG were first analyzed using the interval mapping method, and then, permutation tests were performed to confirm the significant thresholds at linkage group-wide and genome-wide with 1000 replicates at a confidence interval of 95%. According to the reference genome of L. maculatus (ASM402866v1), potential candidate genes were extracted within the ± 50-kb genome regions surrounding these significant SNPs.
Results
2b-RAD Sequencing
A total of 2.26 billion raw reads were produced containing the female parent (9.20 million reads), the male parent (9.20 million reads), and their 333 progenies (averaged 6.74 million reads per progeny). After filtering, 2 billion clean reads were adopted for further analysis. The averaged sequencing depths were 27.42-fold for parents and 20.80-fold for progenies. The averaged mapping rates for all samples were more than 88% (Table 1). The raw read data were submitted to the NCBI Sequence Read Archive (SRA) with Accession Number PRJNA580292.
SNP Filtering and Linkage Map Construction
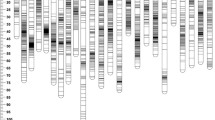
A total of 7,334,449 SNP markers were initially obtained from two parents and 333 progenies. After step-by-step filtering, finally, only 11,360 SNP markers were used to construct the linkage map (Supplementary Table S2). Among these markers, three segregation types were classified, including nn × np, lm × ll, and hk × hk. These two sex-specific maps were primarily constructed and then integrated to produce the sex-averaged linkage map (Fig. 1; Tables 2 and 3). A total of 24 LGs were detected, which was consistent with the haploid chromosome number of L. maculatus (Sha et al. 2003). The total length of the female-specific linkage map was 1929.90 cM with an average marker interval of 0.60 cM. The male-specific map length was 1433.61 cM, and its average marker interval was 0.54 cM. The average ratio of female-to-male genetic length was 1.35:1.

Genetic length and marker distribution of 24 LGs in the linkage maps of Lateolabrax maculatus. a–c The male-specific map, the female-specific map, and the sex-averaged map, respectively. The scaleplate on the left indicates genetic distance (cM as unit). The warmer colors indicate a higher density of markers
After integration, the sex-averaged map comprised 6883 markers spanning 2189.96 cM. The average marker interval was 0.33 cM, which ranged from 0.23 cM (LG10 and LG12) to 0.50 cM (LG20) (Table 3). The details about the sex-averaged map and sex-specific maps are presented in Supplementary Table S3.
QTL Mapping of Body Weight and Body Length
A total of 24 QTLs comprising 318 significant SNPs for growth traits were identified on LG1, LG3, LG5, LG10, LG11, LG15, LG20, and LG21 (Fig. 2; Table 4). The phenotypic variance explained (PVE) values varied from 5.1 to 8.6%, with LOD scores ranging from 3.75 to 6.47. For body weight, 13 QTL regions were detected with 5.6 to 8.6% PVE, and qBW-11.1 was identified as only one genome-wide significant QTL (Fig. 3; Table 4). For body length, 11 QTL regions were mapped to 8 different LGs, with PVE ranging from 5.1 to 6.2% (Table 4). These significant QTLs were further amplified to obtain candidate genes for growth traits of L. maculatus (Fig. 3; Supplementary Table S4).

QTL mapping of growth traits of body weight and body length in Lateolabrax maculatus. At the threshold of p < 0.05 (linkage group-wide), 13 QTLs were identified for body weight, and 11 QTLs were characterized for body length. At the threshold of p < 0.01 (linkage group-wide), 5 QTLs were identified for body weight distributed on LG1, LG10, and LG11, and 4 QTLs for body length were solely identified on LG20. Only one QTL on LG11 for body weight reached the genome-wide significance of p < 0.05

Regional amplification of candidate regions for body weight and body length on LG1 (a), LG10 (b), LG11 (c), and LG20 (d). Genes are extracted in the ± 50-kb genome regions surrounding the significant SNP. Peak marker positions within each candidate region are noted with red arrows. The red dashed line indicates linkage group-wide significance at the 1% level. The black dashed line represents linkage group-wide significance at the 5% level. The blue line indicates genome-wide significance at the 5% level
Candidate Genes Identified for Growth Traits
According to genome annotations of L. maculatus, 30 growth-related candidate genes were identified within the detected candidate QTL regions (Table 5; Fig. 3). The potential functions of the genes involved cell adhesion (col4a3, col4a4, pcdhgc5, and pcdh10); cell proliferation, differentiation, and migration (fgfr4, fgf12a, fgf18, prkca, smyd5, foxq1, foxf2, foxc1a, hes6, gpc1b, gpc5c, gpr55, rab11fip5, and pax3a); cytoskeleton reorganization (diaph1, mylk4a, and mid1ip1l); calcium channel (cacng1a, cacng4a, and cacng5a); and neuromodulation (cadm1a, gfra4b, zdhhc9, nyap2b, sytl4, and per2).
Notably, the fgfr4 gene was determined to have the highest significance for growth traits in our study (Fig. 3), which is reported to be necessary for both bone growth and myogenesis (Cinque et al. 2015, 2016; Lagha et al. 2008; Mok et al. 2014; Zhao et al. 2006). The FGF18/FGFR4 signaling pathway is presented in Fig. 4a and was recently demonstrated to play an important role in chondrocyte autophagy during postnatal bone growth in mammals (Cinque et al. 2015, 2016). In addition, the myogenic programs (proliferation and differentiation) were jointly modulated by Pax3, Fgfr4, and Fgf18 expressions, while muscle regeneration was accomplished by activating the MyoD-Tead2-Fgfr4 pathway in mammals (Fig. 4b) (Lagha et al. 2008, Mok et al. 2014, Zhao et al. 2006). The detailed functional mechanism of the genes is described in “Discussion.”

Schematic diagram showing the function of identified candidate genes for growth, including FGFR4, FGF18, and PAX3, in a bone growth and b myogenesis (modified from Cinque et al. (2015) and Lagha et al. (2008)). a During postnatal bone growth, FGF18 induces chondrocyte autophagy through the VPS34-beclin-1 complex via the activation of FGFR4 and JNK kinase. b PAX3 orchestrates muscle stem cells into the myogenic program by direct activation of MYF5 and FGFR4 expressions. FGF18 affects the transcription of MYOD and PAX3 to regulate myogenesis. The MYOD-TEAD2-FGFR4 pathway is important for effective muscle regeneration
Discussion
A linkage map is recognized as the basis for genomic studies, exploration of QTLs and further MAS breeding. The construction of the first linkage map for economically important fish was performed in 1998 in rainbow trout (Oncorhynchus mykiss) (Young et al. 1998), and along with sequencing technological development, high-resolution genetic maps containing thousands of SNPs have been constructed in more than 30 aquaculture species to date (Zhu et al. 2019). In the present study investigating spotted sea bass, the first high-density linkage map was constructed by sequencing 333 individuals using 2b-RAD technology. A total of 6883 SNP markers were anchored onto 24 linkage groups with an average marker interval of 0.33 cM. The length of the L. maculatus female map was longer than the male map with a ratio of 1.35:1. In species with the XY sex determination system, the female map is usually longer than the male map (Chistiakov et al. 2006). This phenomenon has been reported in several teleosts with XY sex determination system, such as medaka (Oryzias latipes) (Kondo et al. 2001), C. carpio (Zhang et al. 2011), I. punctatus (Li et al. 2015), and C. auratus (Liu et al. 2017).
Genetic markers can be used to directly select promising breeding populations, and QTL mapping provides an effective approach to identify these molecular markers (Zhu et al. 2019). In this study, 24 growth-related QTLs were identified on eight LGs, including 13 QTLs for body weight and 11 QTLs for body length. Notably, 6 QTL intervals were shared in both growth traits (Fig. 2), indicating that a higher phenotypic correlation occurred between body weight and body length and that one trait selection may result in the improvement of another growth trait in the breeding of spotted sea bass (Qiu et al. 2018). Furthermore, 318 growth-related SNPs and 30 candidate genes were provided for further MAS breeding of L. maculatus. These candidate genes serve essential functions in multiple growth-related procedures, such as cell adhesion, cell proliferation, cytoskeleton reorganization, calcium channels, and neuromodulation.
Our results showed that the fgfr4 gene was detected in the candidate region with the highest significance; this gene mediates cell proliferation and cell differentiation and is prominently expressed in avian developing skeletal muscles (Shin and Osborne 2009; Marics et al. 2002). A genome-wide association study (GWAS) also showed that FGFR4 variations may influence human height (Lango Allen et al. 2010). As a ligand of Fgfr4 (Falvella et al. 2009), Fgf18 plays an essential role in chondrogenesis and osteogenesis in mammals (Ohbayashi et al. 2002) and is also regarded as an important regulator in GWAS analysis of growth traits in large yellow croaker (Larimichthys crocea) (Zhou et al. 2019c). In mammals, the FGF18/FGFR4 pathway is identified as a novel effector of FGF signaling via controlling chondrocyte autophagy to maintain postnatal bone growth (Cinque et al. 2015, 2016). Another candidate gene of fgf12a was also identified through QTL analysis. This result suggests that FGF signaling may play an important role in the growth of L. maculatus. Furthermore, Pax3 regulates myogenesis through direct activation of both the Fgfr4 gene and the myogenic determination gene of Myf5 (Lagha et al. 2008); meanwhile, Fgf18 signaling can upregulate another differentiation gene of MyoD and has a potential effect on Pax3 expression (Mok et al. 2014; Mohammed et al. 2017). Moreover, the MyoD gene may activate Fgfr4 expression through the MyoD-Tead2-Fgfr4 pathway to regulate muscle regeneration in mice (Mus musculus) (Zhao et al. 2006). However, the studies of these genes in teleost growth are comparatively limited, with only a few species, such as zebrafish (Danio rerio) (Thisse et al. 1995) and olive flounder (Paralichthys olivaceus) (Jiao et al. 2017), being investigated.
Neuromodulation may be a major factor explaining the higher growth performance of fish MAS breeding (Su et al. 2018). Body weight is determined by a balance between food intake and energy expenditure, which is regulated by multiple neural circuits (Rui 2013). Several neuromodulation genes identified in our study have been reported to exert essential roles in related growth processes. The cadm1a gene regulates M. musculus body weight and energy homeostasis by mediating synaptic assembly, which has also been identified in GWAS analysis of body mass index (BMI) in humans (Locke et al. 2015). The Gfra4 gene is expressed in the neural crest, which is necessary for endocrine cell development in M. musculus (Lindahl et al. 2000). The Zdhhc9 gene plays an important role in dendrite outgrowth and inhibitory synapse formation (Shimell et al. 2019), which is also identified as a vital candidate gene for growth-related QTLs in C. carpio haematopterus (Feng et al. 2018). Additionally, the DIAPH1 protein is expressed in human neuronal precursor cells, and patients with a homozygous loss of the DIAPH1 gene suffer from microcephaly and reduced height and weight (Ercan-Sencicek et al. 2015). The important roles played by neuromodulation in growth control are specifically discussed in the genetic breeding of C. carpio (Su et al. 2018).
Cell adhesion plays important roles in cell-cell communication and the development and maintenance of tissues (Khalili and Ahmad 2015). The candidate genes of col4a3 and col4a4 are detected in the basement membrane of Prochilodus argenteus, which is important for maintaining normal cell adhesion to the extracellular matrix (Thomé et al. 2010). The pcdh10 gene, as a cell adhesion factor, exerts vital effects on paraxial mesoderm development and somitogenesis in D. rerio (Murakami et al. 2006). However, further studies are needed to corroborate the roles of cell adhesion genes in L. maculatus growth regulation. For calcium channels, these candidate gene functions are focused on bone resorption via osteoclasts, which is necessary for skeletal development in teleosts (Witten et al. 2000; Zayzafoon 2006).
Conclusion
In this study, the first high-density linkage map of L. maculatus was constructed, which contained 6883 SNP markers spanning 2189.96 cM. For body weight and body length QTL mapping, 24 significant QTLs, including 318 SNPs and 30 candidate genes, were identified. These genes are involved in cell adhesion, cell proliferation, cytoskeleton reorganization, calcium channels, and neuromodulation. Our results provide a useful framework for determining the genetic basis for growth traits in spotted sea bass and establish a foundation for further molecular-assisted breeding programs for this species.
Data Availability
The sequencing data of this study are deposited in NCBI Sequence Read Archive (SRA) with Accession Number PRJNA580292.
References
Ashton DT, Ritchie PA, Wellenreuther M (2019) High-density linkage map and QTLs for growth in snapper (Chrysophrys auratus). G3 (Bethesda) 9:1027–1035
Cai L-S, Wang L, Song K, Lu K-L, Zhang C-X, Rahimnejad S (2020) Evaluation of protein requirement of spotted seabass (Lateolabrax maculatus) under two temperatures, and the liver transcriptome response to thermal stress. Aquaculture 516:734615
Chen B, Li Y, Peng W, Zhou Z, Shi Y, Pu F, Luo X, Chen L, Xu P (2019a) Chromosome-level assembly of the Chinese seabass (Lateolabrax maculatus) genome. Front Genet 10:275–275
Chen J, Jayachandran M, Xu B, Yu Z (2019b) Sea bass (Lateolabrax maculatus) accelerates wound healing: a transition from inflammation to proliferation. J Ethnopharmacol 236:263–276
Chistiakov DA, Hellemans B, Volckaert FAM (2006) Microsatellites and their genomic distribution, evolution, function and applications: a review with special reference to fish genetics. Aquaculture 255:1–29
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6:80–92
Cinque L, Forrester A, Bartolomeo R, Svelto M, Venditti R, Montefusco S, Polishchuk E, Nusco E, Rossi A, Medina DL, Polishchuk R, De Matteis MA, Settembre C (2015) FGF signalling regulates bone growth through autophagy. Nature 528:272–275
Cinque L, Forrester A, Settembre C (2016) Autophagy gets to the bone. Cell Cycle 15:871–872
Dong C, Jiang P, Zhang J, Li X, Li S, Bai J, Fan J, Xu P (2019) High-density linkage map and mapping for sex and growth-related traits of largemouth bass (Micropterus salmoides). Front Genet 10:960–960
Ercan-Sencicek AG, Jambi S, Franjic D, Nishimura S, Li M, El-Fishawy P, Morgan TM, Sanders SJ, Bilguvar K, Suri M, Johnson MH, Gupta AR, Yuksel Z, Mane S, Grigorenko E, Picciotto M, Alberts AS, Gunel M, Sestan N, State MW (2015) Homozygous loss of DIAPH1 is a novel cause of microcephaly in humans. Eur J Hum Genet 23:165–172
Falvella FS, Frullanti E, Galvan A, Spinola M, Noci S, De Cecco L, Nosotti M, Santambrogio L, Incarbone M, Alloisio M, Calabrò E, Pastorino U, Skaug V, Haugen A, Taioli E, Dragani TA (2009) FGFR4 Gly388Arg polymorphism may affect the clinical stage of patients with lung cancer by modulating the transcriptional profile of normal lung. Int J Cancer 124:2880–2885
Fan H, Wang L, Wen H, Wang K, Qi X, Li J, He F, Li Y (2019a) Genome-wide identification and characterization of toll-like receptor genes in spotted sea bass (Lateolabrax maculatus) and their involvement in the host immune response to Vibrio harveyi infection. Fish Shellfish Immunol 92:782–791
Fan H, Zhou Y, Wen H, Zhang X, Zhang K, Qi X, Xu P, Li Y (2019b) Genome-wide identification and characterization of glucose transporter (glut) genes in spotted sea bass (Lateolabrax maculatus) and their regulated hepatic expression during short-term starvation. Comp Biochem Physiol Part D Genomics Proteomics 30:217–229
Feng X, Yu X, Fu B, Wang X, Liu H, Pang M, Tong J (2018) A high-resolution genetic linkage map and QTL fine mapping for growth-related traits and sex in the Yangtze River common carp (Cyprinus carpio haematopterus). BMC Genomics 19:230
Fu X, Dou J, Mao J, Su H, Jiao W, Zhang L, Hu X, Huang X, Wang S, Bao Z (2013) RADtyping: an integrated package for accurate de novo codominant and dominant RAD genotyping in mapping populations. PLoS One 8:e79960
Fu B, Liu H, Yu X, Tong J (2016) A high-density genetic map and growth related QTL mapping in bighead carp (Hypophthalmichthys nobilis). Sci Rep 6:28679–28679
Guo J, Li C, Teng T, Shen F, Chen Y, Wang Y, Pan C, Ling Q (2018) Construction of the first high-density genetic linkage map of pikeperch (Sander lucioperca) using specific length amplified fragment (SLAF) sequencing and QTL analysis of growth-related traits. Aquaculture 497:299–305
Huang W, Cheng C, Liu J, Zhang X, Ren C, Jiang X, Chen T, Cheng K, Li H, Hu C (2020) Fine mapping of the high-pH tolerance and growth trait-related quantitative trait loci (QTLs) and identification of the candidate genes in Pacific white shrimp (Litopenaeus vannamei). Mar Biotechnol 22:1–18
Jiang DL, Gu XH, Li BJ, Zhu ZX, Qin H, Meng ZN, Lin HR, Xia JH (2019) Identifying a long QTL cluster across chrLG18 associated with salt tolerance in tilapia using GWAS and QTL-seq. Mar Biotechnol (NY) 21:250–261
Jiao S, Wu Z, Tan X, Sui Y, Wang L, You F (2017) Characterization of pax3a and pax3b genes in artificially induced polyploid and gynogenetic olive flounder (Paralichthys olivaceus) during embryogenesis. Fish Physiol Biochem 43:385–395
Khalili AA, Ahmad MR (2015) A review of cell adhesion studies for biomedical and biological applications. Int J Mol Sci 16:18149–18184
Kondo M, Nagao E, Mitani H, Shima A (2001) Differences in recombination frequencies during female and male meioses of the sex chromosomes of the medaka, Oryzias latipes. Genet Res 78:23–30
Kong S, Ke Q, Chen L, Zhou Z, Pu F, Zhao J, Bai H, Peng W, Xu P (2019) Constructing a high-density genetic linkage map for large yellow croaker (Larimichthys crocea) and mapping resistance trait against ciliate parasite Cryptocaryon irritans. Mar Biotechnol 21:262–275
Lagha M, Sato T, Bajard L, Daubas P, Esner M, Montarras D, Relaix F, Buckingham M (2008) Regulation of skeletal muscle stem cell behavior by Pax3 and Pax7. Cold Spring Harb Symp Quant Biol 73:307–315
Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S, Ferreira T, Wood AR, Weyant RJ, Segrè AV, Speliotes EK, Wheeler E, Soranzo N, Park J-H, Yang J, Gudbjartsson D, Heard-Costa NL, Randall JC, Qi L, Vernon Smith A, Mägi R, Pastinen T, Liang L, Heid IM, Ja L, Thorleifsson G, Winkler TW, Goddard ME, Sin Lo K, Palmer C, Workalemahu T, Aulchenko YS, Johansson A, Zillikens MC, Feitosa MF, Esko T, Johnson T, Ketkar S, Kraft P, Mangino M, Prokopenko I, Absher D, Albrecht E, Ernst F, Glazer NL, Hayward C, Hottenga J-J, Jacobs KB, Knowles JW, Kutalik Z, Monda KL, Polasek O, Preuss M, Rayner NW, Robertson NR, Steinthorsdottir V, Tyrer JP, Voight BF, Wiklund F, Xu J, Zhao JH, Nyholt DR, Pellikka N, Perola M, Perry JRB, Surakka I, Tammesoo M-L, Altmaier EL, Amin N, Aspelund T, Bhangale T, Boucher G, Chasman DI, Chen C, Coin L, Cooper MN, Dixon AL, Gibson Q, Grundberg E, Hao K, Juhani Junttila M, Kaplan LM, Kettunen J, König IR, Kwan T, Lawrence RW, Levinson DF, Lorentzon M, McKnight B, Morris AP, Müller M, Suh Ngwa J, Purcell S, Rafelt S, Salem RM, Salvi E et al (2010) Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467:832–838
Li Y, Liu S, Qin Z, Waldbieser G, Wang R, Sun L, Bao L, Danzmann RG, Dunham R, Liu Z (2015) Construction of a high-density, high-resolution genetic map and its integration with BAC-based physical map in channel catfish. DNA Res 22:39–52
Li HL, Gu XH, Li BJ, Chen CH, Lin HR, Xia JH (2017) Genome-wide QTL analysis identified significant associations between hypoxia tolerance and mutations in the GPR132 and ABCG4 genes in Nile tilapia. Mar Biotechnol (NY) 19:441–453
Li BJ, Zhu ZX, Gu XH, Lin HR, Xia JH (2019a) QTL mapping for red blotches in Malaysia red tilapia (Oreochromis spp.). Mar Biotechnol (NY) 21:384–395
Li Q, Wen H, Li Y, Zhang Z, Zhou Y, Qi X (2019b) Evidence for the direct effect of the NPFF peptide on the expression of feeding-related factors in spotted sea bass (Lateolabrax maculatus). Front Endocrinol 10:545
Lindahl M, Timmusk T, Rossi J, Saarma M, Airaksinen MS (2000) Expression and alternative splicing of mouse Gfra4 suggest roles in endocrine cell development. Mol Cell Neurosci 15:522–533
Liu H, Fu B, Pang M, Feng X, Yu X, Tong J (2017) A high-density genetic linkage map and QTL fine mapping for body weight in Crucian carp (Carassius auratus) using 2b-RAD sequencing. G3 (Bethesda) 7:2473–2487
Liu Y, Wen H, Qi X, Zhang X, Zhang K, Fan H, Tian Y, Hu Y, Li Y (2019) Genome-wide identification of the Na(+)/H(+) exchanger gene family in Lateolabrax maculatus and its involvement in salinity regulation. Comp Biochem Physiol Part D Genomics Proteomics 29:286–298
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, Croteau-Chonka DC, Esko T, Fall T, Ferreira T, Gustafsson S, Kutalik Z, Ja L, Mägi R, Randall JC, Winkler TW, Wood AR, Workalemahu T, Faul JD, Smith JA, Zhao JH, Zhao W, Chen J, Fehrmann R, Hedman ÅK, Karjalainen J, Schmidt EM, Absher D, Amin N, Anderson D, Beekman M, Bolton JL, Bragg-Gresham JL, Buyske S, Demirkan A, Deng G, Ehret GB, Feenstra B, Feitosa MF, Fischer K, Goel A, Gong J, Jackson AU, Kanoni S, Kleber ME, Kristiansson K, Lim U, Lotay V, Mangino M, Leach IM, Medina-Gomez C, Medland SE, Nalls MA, Palmer CD, Pasko D, Pechlivanis S, Peters MJ, Prokopenko I, Shungin D, Stančáková A, Strawbridge RJ, Sung YJ, Tanaka T, Teumer A, Trompet S, van der Laan SW, van Setten J, Van Vliet-Ostaptchouk JV, Wang Z, Yengo L, Zhang W, Isaacs A, Albrecht E, Ärnlöv J, Arscott GM, Attwood AP, Bandinelli S, Barrett A, Bas IN, Bellis C, Bennett AJ, Berne C, Blagieva R, Blüher M, Böhringer S, Bonnycastle LL, Böttcher Y, Boyd HA, Bruinenberg M, Caspersen IH, Chen Y-DI, Clarke R, Daw EW, de Craen AJM, Delgado G, Dimitriou M et al (2015) Genetic studies of body mass index yield new insights for obesity biology. Nature 518:197–206
Marics I, Padilla F, Guillemot JF, Scaal M, Marcelle C (2002) FGFR4 signaling is a necessary step in limb muscle differentiation. Development 129:4559–4569
Mohammed RH, Anderton H, Brameld JM, Sweetman D (2017) Effects of insulin like growth factors on early embryonic chick limb myogenesis. PLoS One 12:e0185775–e0185775
MOA (Ministry of Agriculture of the People’s Republic of China) (2018) China fishery statistical yearbook. In: Department of fishery of the Ministry of Agriculture. China Agricultural Press, Beijing
Mok GF, Cardenas R, Anderton H, Campbell K, Sweetman D (2014) Interactions between FGF18 and retinoic acid regulate differentiation of chick embryo limb myoblasts. Dev Biol 396:214–223
Murakami T, Hijikata T, Matsukawa M, Ishikawa H, Yorifuji H (2006) Zebrafish protocadherin 10 is involved in paraxial mesoderm development and somitogenesis. Dev Dyn 235:506–514
Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N, Takada S (2002) FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev 16:870–879
Peng W, Xu J, Zhang Y, Feng J, Dong C, Jiang L, Feng J, Chen B, Gong Y, Chen L, Xu P (2016) An ultra-high density linkage map and QTL mapping for sex and growth-related traits of common carp (Cyprinus carpio). Sci Rep 6:26693–26693
Qiu C, Han Z, Li W, Ye K, Xie Y, Wang Z (2018) A high-density genetic linkage map and QTL mapping for growth and sex of yellow drum (Nibea albiflora). Sci Rep 8:17271–17271
Rui L (2013) Brain regulation of energy balance and body weight. Rev Endocr Metab Disord 14:387–407
Sha Z, Chen S, Ye H, Xu M, Liu Y, Ji X, Tang Q (2003) Comparison of several chromosome preparation methods in sea perch Lateolabrax japonicus. J Fish China 10:469–473. (In China)
Shao C, Li C, Wang N, Qin Y, Xu W, Liu Q, Zhou Q, Zhao Y, Li X, Liu S, Chen X, Mahboob S, Liu X, Chen S (2018) Chromosome-level genome assembly of the spotted sea bass, Lateolabrax maculatus. Giga Sci 7:giy114
Shen Y, He Y, Bi Y, Chen J, Zhao Z, Li J, Chen X (2019) Transcriptome analysis of gill from Lateolabrax maculatus and aqp3 gene expression. Aquac Fish 6:247–254
Shi Y, Zhou Z, Liu B, Kong S, Chen B, Bai H, Li L, Pu F, Xu P (2020) Construction of a high-density genetic linkage map and QTL mapping for growth-related traits in Takifugu bimaculatus. Mar Biotechnol 22:130–144
Shimell JJ, Shah BS, Cain SM, Thouta S, Kuhlmann N, Tatarnikov I, Jovellar DB, Brigidi GS, Kass J, Milnerwood AJ (2019) The X-linked intellectual disability gene Zdhhc9 is essential for dendrite outgrowth and inhibitory synapse formation. Cell Rep 29:2422–2437. e8
Shin DJ, Osborne TF (2009) FGF15/FGFR4 integrates growth factor signaling with hepatic bile acid metabolism and insulin action. J Biol Chem 284:11110–11120
Su S, Li H, Du F, Zhang C, Li X, Jing X, Liu L, Li Z, Yang X, Xu P, Yuan X, Zhu J, Bouzoualegh R (2018) Combined QTL and genome scan analyses with the help of 2b-RAD identify growth-associated genetic markers in a new fast-growing carp strain. Front Genet 9:592
Sun C, Niu Y, Ye X, Dong J, Hu W, Zeng Q, Chen Z, Tian Y, Zhang J, Lu M (2017) Construction of a high-density linkage map and mapping of sex determination and growth-related loci in the mandarin fish (Siniperca chuatsi). BMC Genomics 18:446–446
Thisse B, Thisse C, Weston JA (1995) Novel FGF receptor (Z-FGFR4) is dynamically expressed in mesoderm and neurectoderm during early zebrafish embryogenesis. Dev Dyn 203:377–391
Thomé R, Dos Santos HB, Sato Y, Rizzo E, Bazzoli N (2010) Distribution of laminin β2, collagen type IV, fibronectin and MMP-9 in ovaries of the teleost fish. J Mol Histol 41:215–224
Tian Y, Wen H, Qi X, Mao X, Shi Z, Li J, He F, Yang W, Zhang X, Li Y (2019a) Analysis of apolipoprotein multigene family in spotted sea bass (Lateolabrax maculatus) and their expression profiles in response to Vibrio harveyi infection. Fish Shellfish Immunol 92:111–118
Tian Y, Wen H, Qi X, Zhang X, Li Y (2019b) Identification of mapk gene family in Lateolabrax maculatus and their expression profiles in response to hypoxia and salinity challenges. Gene 684:20–29
Tian Y, Wen H, Qi X, Zhang X, Liu S, Li B, Sun Y, Li J, He F, Yang W (2019c) Characterization of full-length transcriptome sequences and splice variants of Lateolabrax maculatus by single-molecule long-read sequencing and their involvement in salinity regulation. Front Genet 10:1126
Van Ooijen J (2006) Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen, Netherlands
Van Ooijen J, Kyazma B (2009) MapQTL 6. Software for the mapping of quantitative trait loci in experimental populations of diploid species. Kyazma BV, Wageningen
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang L, Wan Z, Bai B, Qing H, Elaine C, May L, Yan P, Fei W, Peng L, Feng L, Fei S, Lin G, Ye B, Yue GH (2015) Construction of a high-density linkage map and fine mapping of QTL for growth in Asian seabass. Sci Rep 5:16358
Wang J, Xue D-X, Zhang B-D, Li Y-L, Liu B-J, Liu J-X (2016a) Genome-wide SNP discovery, genotyping and their preliminary applications for population genetic inference in spotted sea bass (Lateolabrax maculatus). PLoS One 11:e0157809
Wang S, Liu P, Lv J, Li Y, Cheng T, Zhang L, Xia Y, Sun H, Hu X, Bao Z (2016b) Serial sequencing of isolength RAD tags for cost-efficient genome-wide profiling of genetic and epigenetic variations. Nat Protoc 11:2189–2200
Wang W, Ma C, Chen W, Zhang H, Kang W, Ni Y, Ma L (2017) Population genetic diversity of Chinese sea bass (Lateolabrax maculatus) from southeast coastal regions of China based on mitochondrial COI gene sequences. Biochem Syst Ecol 71:114–120
Wang H, Wen H, Li Y, Zhang K, Liu Y (2018) Evaluation of potential reference genes for quantitative RT-PCR analysis in spotted sea bass (Lateolabrax maculatus) under normal and salinity stress conditions. PeerJ 6:e5631–e5631
Wang L, Chua E, Sun F, Wan ZY, Ye B, Pang H, Wen Y, Yue GH (2019a) Mapping and validating QTL for fatty acid compositions and growth traits in Asian seabass. Mar Biotechnol 21:643–654
Wang L, Xie N, Shen Y, Ye B, Yue GH, Feng X (2019b) Constructing high-density genetic maps and developing sexing markers in northern snakehead (Channa argus). Mar Biotechnol (NY) 21:348–358
Wang W, Tan S, Luo J, Shi H, Zhou T, Yang Y, Jin Y, Wang X, Niu D, Yuan Z (2019c) GWAS analysis indicated importance of NF-κB signaling pathway in host resistance against motile Aeromonas septicemia disease in catfish. Mar Biotechnol 21:335–347
Witten P, Villwock W, Peters N, Hall B (2000) Bone resorption and bone remodelling in juvenile carp, Cyprinus carpio L. J Appl Ichthyol 16:254–261
Wu L, Yang Y, Li B, Huang W, Wang X, Liu X, Meng Z, Xia J (2019) First genome-wide association analysis for growth traits in the largest coral reef-dwelling bony fishes, the Giant grouper (Epinephelus lanceolatus). Mar Biotechnol 21:707–717
Young WP, Wheeler PA, Coryell VH, Keim P, Thorgaard GH (1998) A detailed linkage map of rainbow trout produced using doubled haploids. Genetics 148:839–850
Zayzafoon M (2006) Calcium/calmodulin signaling controls osteoblast growth and differentiation. J Cell Biochem 97:56–70
Zhan A, Bao Z, Lu WEI, Hu X, Peng WEI, Wang M, Hu J (2007) Development and characterization of 45 novel microsatellite markers for sea cucumber (Apostichopus japonicus). Mol Ecol Notes 7:1345–1348
Zhang Y, Xu P, Lu C, Kuang Y, Zhang X, Cao D, Li C, Chang Y, Hou N, Li H, Wang S, Sun X (2011) Genetic linkage mapping and analysis of muscle fiber-related QTLs in common carp (Cyprinus carpio L.). Mar Biotechnol (NY) 13:376–392
Zhang K-Q, Hou Z-S, Wen H-S, Li Y, Qi X, Li W-J, Tao Y-X (2019a) Melanocortin-4 receptor in spotted sea bass, Lateolabrax maculatus: cloning, tissue distribution, physiology, and pharmacology. Front Endocrinol 10:705
Zhang K-q, Wen H-s, Li J-f, Qi X, H-y F, X-y Z, Tian Y, Liu Y, Wang H-l, Li Y (2019b) 14-3-3 gene family in spotted sea bass (Lateolabrax maculatus): genome-wide identification, phylogenetic analysis and expression profiles after salinity stress. Comp Biochem Physiol A Mol Integr Physiol 235:1–11
Zhang S, Zhang X, Chen X, Xu T, Wang M, Qin Q, Zhong L, Jiang H, Zhu X, Liu H, Shao J, Zhu Z, Shi Q, Bian W, You X (2019c) Construction of a high-density linkage map and QTL fine mapping for growth- and sex-related traits in channel catfish (Ictalurus punctatus). Front Genet 10:251
Zhang X, Wen H, Qi X, Zhang K, Liu Y, Fan H, Yu P, Tian Y, Li Y (2019d) Na+-K+-ATPase and nka genes in spotted sea bass (Lateolabrax maculatus) and their involvement in salinity adaptation. Comp Biochem Physiol A Mol Integr Physiol 235:69–81
Zhang Z, Wen H, Li Y, Li Q, Li W, Zhou Y, Wang L, Liu Y, Lyu L, Qi X (2019e) TAC3 gene products regulate brain and digestive system gene expression in the spotted sea bass (Lateolabrax maculatus). Front Endocrinol 10:556
Zhang Y, Liu Z, Li H (2020) Genomic prediction of columnaris disease resistance in catfish. Mar Biotechnol (NY) 22:145–151
Zhao P, Caretti G, Mitchell S, McKeehan WL, Boskey AL, Pachman LM, Sartorelli V, Hoffman EP (2006) Fgfr4 is required for effective muscle regeneration in vivo. Delineation of a MyoD-Tead2-Fgfr4 transcriptional pathway. J Biol Chem 281:429–438
Zhou C, Lin H, Huang Z, Wang J, Wang Y, Yu W (2019a) Transcriptome analysis reveals differential gene expression in Lateolabrax maculatus following waterborne Zn exposure. Aquac Rep 15:100229
Zhou Y, Qi X, Wen H, Zhang K, Zhang X, Li J, Li Y, Fan H (2019b) Identification, expression analysis, and functional characterization of motilin and its receptor in spotted sea bass (Lateolabrax maculatus). Gen Comp Endocrinol 277:38–48
Zhou Z, Han K, Wu Y, Bai H, Ke Q, Pu F, Wang Y, Xu P (2019c) Genome-wide association study of growth and body-shape-related traits in large yellow croaker (Larimichthys crocea) using ddRAD sequencing. Mar Biotechnol 21:655–670
Zhou Y, Liu H, Wang X, Fu B, Yu X, Tong J (2020) QTL fine mapping for sex determination region in bighead carp (Hypophthalmichthys nobilis) and comparison with silver carp (Hypophthalmichthys molitrix). Mar Biotechnol (NY) 22:41–53
Zhu C, Liu H, Pan Z, Chang G, Wang H, Wu N, Ding H, Yu X (2019) Construction of a high-density genetic linkage map and QTL mapping for growth traits in Pseudobagrus ussuriensis. Aquaculture 511:734213
Acknowledgments
We thank Luoluo Chen, Shuxian Wu, Yangyang Zhou, and Bingyu Li for sample collection.
Funding
This project was supported by the National Key R&D Program of China (2018YFD0900101) and the China Agriculture Research System (CARS-47).
Author information
Authors and Affiliations
Contributions
Yang Liu and Haolong Wang contribute equally as first authors. Yun Li, Haishen Wen, Qingli Gong, and Xin Qi conceived and supervised the study. Meizhao Zhang, Jifang Li, Feng He, and Kaiqiang Zhang worked on the mapping family construction. Yang Liu, Haolong Wang, and Yanbo Hu collected samples. Yang Liu, Haolong Wang, and Yue Shi performed the analysis and designed the charts and tables. Yang Liu wrote the manuscript. Yun Li revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
All experimental procedures involving the fish were conducted in accordance with approved guidelines of the respective Animal Research and Ethics Committees of Ocean University of China (Permit Number: 20141201). The present study did not include endangered or protected species.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, Y., Wang, H., Wen, H. et al. First High-Density Linkage Map and QTL Fine Mapping for Growth-Related Traits of Spotted Sea bass (Lateolabrax maculatus). Mar Biotechnol 22, 526–538 (2020). https://doi.org/10.1007/s10126-020-09973-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-020-09973-4