
Abstract
Schizothorax prenanti (S. prenanti) is an indigenous fish species and is popularly cultured in southwestern China. In recent years, intensive farming of S. prenanti and water quality deterioration has increased the susceptibility of this fish to various pathogens, including Aeromonas hydrophila (A. hydrophila), which has caused severe damage to S. prenanti production. However, the understanding of molecular immune response of S. prenanti to A. hydrophila infection is still lacking. In order to better comprehend the S. prenanti time series immune response process against A. hydrophila, we conducted the first transcriptomic comparison in S. prenanti spleen at 4, 24, and 48 h after the infection challenge of A. hydrophila against their control counterparts. In total, 628 million clean reads were obtained from 18 libraries and assembled into 262,745 transcripts. After eliminating sequence redundancy, 69,373 unigenes with an average length of 1476 bp were obtained. Comparative analysis revealed 1890 unigenes with significantly differential expression, including 172, 455, 589 upregulated and 27, 676, 551 unigenes downregulated genes for 4, 24, and 48 h post-infection, respectively. Differentially expressed genes (DEGs) were validated using qPCR for 15 randomly selected genes. Enrichment and pathway analysis of DEGs was carried out to understand the functions of the immune-related genes. Our results revealed that many important functional genes relating to complement and coagulation cascades, chemokine signaling pathway, toll-like receptor signaling pathway, NOD-like receptor signaling pathway and leukocyte transendothelial migration were regulated during the infection of A. hydrophila, and the expression of those genes reflected the transcriptome profiles during the challenging stages.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Schizothorax prenanti (Tchang) (Cypriniformes: Cyprinidae: Schizothoracinae), also named Ya-fish, is an indigenous herbivorous freshwater fish species in China, and an important commercial cold-water fish commonly distributed in the upstream of the Yangtze River and its tributaries in China. It is a benthic species inhabiting montane rivers with relatively low temperatures and a rapid current (Ding 1994). This species is a reputed cold-water aquaculture species in west China because of its good meat quality, wonderful flavor, nutritional value, and high commercial value (Ding 1994). The rapid expansion and intensification of S. prenanti aquaculture have led to a series of problems, including the occurrence of various diseases that have limited the sustainable development of the industry. Outbreaks of disease associated with various pathogens, such as Aeromonas hydrophila (A. hydrophila), have caused a high infection ratio, leading to reduced production and considerable economic losses (Du et al. 2003, 2011; Geng et al. 2012). Meanwhile, the wild resources of S. prenanti are rapidly decreasing because of over-fishing, water pollution, and construction of hydropower stations (Song et al. 2006; Liang et al. 2011; Zhang et al. 2015a). In order to formulate the optimal therapeutic regimens for the diseases and to protect germplasm resources of the fish species, there is a great demand to study the molecular mechanisms of immune defense for S. prenanti. Previous reports in S. prenanti mainly focused on the biological functions of single genes, such as agouti-related protein (AgRP) (Wei et al. 2013b), melanocortin 4 receptor (MC4R) (Wei et al. 2013c), ghrelin (Wei et al. 2013a), peptide YY (Yuan et al. 2014b), leptin, and cholecystokinin (Yuan et al. 2014a). The systematic studies of genomic data for S. prenanti are still limited (Luo et al. 2016; Du et al. 2017).
The genus Aeromonas, belonging to the family Aeromonadaceae, is not only an important disease-causing pathogen for fish and other cold-blooded species but also an etiologic agent responsible for a variety of infectious complications in humans (Joseph and Carnahan 1994; Janda and Abbott 2010). A. hydrophila is a gram-negative motile bacillus widely distributed in aquatic environments (Colwell et al. 1986; Janda and Abbott 2010). As an opportunistic bacterial pathogen, A. hydrophila causes outbreaks of diseases in fish, amphibians, reptiles, and humans (Singh et al. 2013). In the aquaculture industry, A. hydrophila has been related to major fish die-offs around the globe over the past decade, resulting in enormous economic losses (Janda and Abbott 2010). Similar to other fish species, A. hydrophila is a serious threat for cultured S. prenanti, because of its ubiquitous presence in the aquatic environment. It causes several disease symptoms including skin ulcer, swelling of tissues, and hemorrhagic septicemia, and so on (Du et al. 2011). Although several methods have been developed to alleviate symptoms and to lower the mortality rate, it is difficult to cure the disease. More effective measures against A. hydrophila infection in fish are needed. Therefore, it is critical to understand the immune molecular mechanisms against A. hydrophila infection in S. prenanti. Several researches have been conducted on the etiology, histopathology, and serology of A. hydrophila in S. prenanti (Du et al. 2003, 2011; Ye et al. 2014). However, analysis of transcriptomes in S. prenanti under A. hydrophila infection has not been reported yet. Although S. prenanti do not have genomic resources available, transcriptome analysis by RNA-seq can still be applied, which provides us a high-throughput tool to probe the time-series gene expression pattern and to reveal the underlying molecular mechanism in the cell upon infection (Song et al. 2017a). At present, RNA-seq technologies are being applied to more and more aquatic animals, including Pseudosciaena crocea (Mu et al. 2010), Ctenopharyngodon idellus (Yang et al. 2016), Portunus trituberculatus (Li et al. 2017), Crassostrea gigas (Zhu et al. 2016), and Clarias macrocephalus (Chatchaiphan et al. 2017).
The spleen is not only an important lymphohematopoietic organ associated with immune responses in fish but also a main target organ for pathogenic bacteria infection. The time-series gene expression pattern of the S. prenanti spleen can be expected to highlight a mass of genes for fish immune responses and would contribute to understanding host-pathogen interactions. So far, many researches have been conducted to reveal gene expression profiles induced by A. hydrophila infection in fish species including C. idellus (Yang et al. 2016), Cyprinus carpio (Jiang et al. 2016), Labeo rohita (Robinson et al. 2012), P. crocea (Mu et al. 2010), and Megalobrama amblycephala (Tran et al. 2015). However, the report for S. prenanti in response to A. hydrophila infection is still lacking. In order to understand S. prenanti’s response to A. hydrophila at the transcriptomic level, we examined the gene expression profiles of S. prenanti spleen at 4, 24, and 48 h post-infection. By comparing the time-series gene expression pattern between challenged and controlled groups, we discovered that many important immune-related and disease resistance genes were differentially expressed after the challenge. This result provides an important resource for understanding the response to A. hydrophila in S. prenanti and constitutes the basis of future studies aiming at biological function analysis of resistance-related genes and molecular assisted breeding of S. prenanti.
Materials and Methods
Fish and Bacterial Challenge
One hundred and eighty-one-year-old S. prenanti (105 g mean weight) were obtained from a culture farm in Meishan, Sichuan Province, China. The fish were reared at the Fisheries Breeding and Healthy Cultivation Research Centre at 19 ± 1 °C in aerated water for two weeks before bacterial challenge. Fish were randomly divided into six aquaria of which three aquaria were designated control (4, 24, and 48 h) and the other three aquaria were designated challenge groups (4, 24, and 48 h), with 30 fish in each aquarium. The fish were fed a commercial pelleted feed (Sichuan Giastar Group; particle diameter 2 mm) twice a day.
The bacteria A. hydrophila, provided by the Aquatic Microbiology Laboratory, Southwest University, were isolated from naturally diseased fish. Healthy fish were artificially infected with isolated bacteria, and the symptom of infected fish was similar to those of the naturally diseased fish. The bacteria were further confirmed to be A. hydrophila by means of its physiological and biochemical indexes, and 16S rDNA sequence. We conducted a challenge trial to determine the median lethal concentration (LD50), using five concentrations of A. hydrophila (control, 106, 107, 108, and 109 cfu/mL) to be injected intraperitoneally. The LD50 was 2.5 × 107 cfu/mL. The fish used in both the challenge and control groups were intraperitoneally injected with equal volumes (0.2 mL) of a bacterial suspension at the LD50 and 0.65% sterile physiological saline, respectively. The experimental protocols used here were approved by the institutional animal care and use committee of Southwest University under protocol no. 0140-269.
Tissue Collection
Spleen tissues were sampled at the 4, 24, and 48 h time points post-infection. At each time point, 18 fish from both control and challenge were randomly selected and divided into three replicate pools (six fish each) respectively. The fish were euthanized with tricaine methanesulfonate (MS-222) at 300 mg/L (buffered with sodium bicarbonate) before tissues were collected. Spleen tissues were stored in 1 mL Sample Protector for RNA (TaKaRa, Dalian, China) at 4 °C overnight. The samples were then transferred to a − 80 °C ultra-low freezer until preparation of RNA.
In order to observe the change of histopathology, five fish were collected from each of the control and treatment aquaria at each time point and euthanized with MS-222 (300 mg/L). Spleen tissues were sampled separately, immediately fixed in Bouin’s fluid at 4 °C for 12 h, and then stored in 70% ethanol until being processed for paraffin embedding. Thin sections (3 μm) were stained with H&E for microscopic observation.
RNA Extraction, Sample Preparation for RNA-Seq, and Sequencing
RNA extraction, preparation, and sequencing methods used in previous work were employed in this study (Luo et al. 2016). Briefly, Total RNA was extracted using TRIzol reagent (Invitrogen, USA) and incubated for 1 h at 37 °C with 10 units of DNase I (TaKaRa, Dalian, China) to eliminate genomic DNA. RNA quality and quantity were analyzed using a BioAnalyzer 2100 (Agilent Technology, Santa Clara, CA) and NanoDrop 2000 spectrophotometer (Infinigen Biotechnology Inc., City of Industry, CA), respectively. Poly(A)+ RNA was purified with oligo(dT) magnetic beads and fragmented into short sequences. First-strand cDNA was synthesized using random hexamer primers and Superscript III (Invitrogen, Carlsbad, CA, USA); this was followed by second-strand cDNA synthesis, end repair, and adaptor ligation. Finally, libraries with insert lengths of ~280 bp were created by PCR amplification and purification. The 18 libraries were barcoded and prepared for sequencing by the Beijing Berry Genomics Co., Ltd., using the manufacturer’s instructions. Sequencing was conducted on an Illumina HiSeq 2500 as 125 bp paired-end reads (Illumina Inc., San Diego, CA, USA).
Transcriptome De Novo Assembly and Annotation
Raw image data were transformed by base calling into sequence data. The quality of sequences was assessed using FastQC V0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Quality filtering was conducted on read pairs using the high-throughput quality control (HTQC) toolkit V0.90.8 (Yang et al. 2013), following quality filtering criteria identified in the previous researches (Luo et al. 2016). The resulting cleaned reads were used in the downstream analyses. The Trinity package V2.3.2 was used for transcript assembly using default parameters (Grabherr et al. 2011). The assembled transcripts were then processed through the Evigene package (http://arthropods.eugenes.org/EvidentialGene/) to eliminate sequence redundancy, with default parameters (Nakasugi et al. 2014). The resulting transcripts that showed significant similarities (> 90%) were then clustered and the longest transcripts for each group were selected as representative unigenes, which were then used for functional annotation. Sequence-length statistics of the assembled transcriptome were performed using our own Python scripts. For gene annotation, BLAST V2.6.0 package (with an E-value threshold of 1 × 10−5) of all unigenes was performed in the National Center for Biotechnology Information (NCBI) non-redundant nucleotide sequence (nt) and non-redundant protein (nr) databases (Altschul et al. 1997). Transcripts were further annotated using the Swiss-Prot, Gene Ontology (GO), EC (Enzyme Code), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases with Blast2GO (Conesa et al. 2005).
Analysis of Differentially Expressed Genes
To estimate the expression profiles for genes, the clean reads were mapped to the transcriptome data of the 18 cDNA libraries using RSEM V1.3.0 (Li and Dewey 2011). The expression levels of unigenes were evaluated by FPKM (the fragments per kilo bases per million mapped reads) (Mortazavi et al. 2008). The identification of differentially expressed genes (DEGs) was performed by DEGseq V3.4 based on TMM normalized counts (Wang et al. 2010). The DEGs were regarded as significant by the following condition: the false discovery rate (FDR) ≤ 0.05 and the absolute value of Log2 (fold change) ≥ 2. Especially, if the FPKM of a gene at any time point was at least twofold of the FPKM value at the previous time point during the course of A. hydrophila challenge, it was been deemed a continuously upregulated gene. DEGs were further annotated by GO and KEGG pathway analyses. GO and KEGG enrichment analyses were performed using the hypergeometric distribution test, with the phyper function in the R software package V3.0.3 (http://www.rproject.org/).
Quantitative Real-Time PCR Analysis
To validate the reliability of the transcriptome data, reverse transcription quantitative real-time PCR (qRT-PCR) analysis was performed on randomly selected 15 unigenes related to immunity. Spleen tissues were sampled from 18 S. prenanti (three biological replicate sample pools (six fish for each pool)) at each time point, which were used in the sequencing libraries preparation. Total RNA was extracted using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions. Reverse transcription was performed using total RNA (4 μg) with GoScript™ Reverse Transcription kit (Promega, USA) following the manufacture’s protocol. Primer set was designed based on RNA-seq unigene sequences by Primer Premier 5.0 and synthesized by BGI tech (Beijing) Co., Ltd. β-actin was used as the reference gene. The qRT-PCR was performed with SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) on a StepOnePlus™ thermocycler (Applied Biosystems, USA). The two-step reaction procedure was as follows: 95 °C for 30 s, 95 °C for 5 s, 60 °C for 30 s, 40 cycles, followed by a dissociation stage. All samples were performed in triplicate and relative gene expression levels were calculated using the 2−△△CT method (Livak and Schmittgen 2001).
Results
Early Symptoms of A. hydrophila Challenge
The artificial challenge with A. hydrophila resulted in noticeable and typical symptoms around 24 h after injection. External symptoms included stagnancy, skin darkening, circling listlessly, increased mucus secretion, and faster respiration. Fish began to die at 48 h post-infection. The majority of infected fish exhibited reddening and hemorrhage on the body, especially around the mouth, chin, cephalothorax, gill, and fins (Fig. 1(A)). Several fish showed ulceration, exophthalmia, and periorbital and intra-ocular hemorrhage (Fig. 1(A)). Ascites, splenomegalia, and hepatomegalia were observed during the dissection. No mortality and apparent symptoms were observed in the control group. In addition, cellular depletion and hemorrhage were observed in the spleen biopsy (Fig. 1(B)). There were no distinct histological changes in any spleen samples from the control group.

Symptoms of body and spleen of S. prenanti. The characteristic reddening, hemorrhage, and necrosis symptom was visible on the infected S. prenanti body surface and spleen
Sequencing and De Novo Assembly of the Spleen Transcriptome
Sequencing of the 18 cDNA libraries obtained a total of 632.94 million raw reads with an average read length of 125 bp, resulting in a total of 79.12 Gb (Table 1). After removal of adaptor and primer, ploy-N-containing reads and low-quality reads, about 628.37 (99.28%) million clean reads with an average read length of 120.38 bp were obtained (Table 1). All clean reads in this article are available in the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/) under Accession number SRR5469042 to SRR5469059.
All clean reads were used for the de novo transcriptome assembly with the Trinity package (Grabherr et al. 2011) with default parameters. As a result, we obtained 262,745 transcripts ranging from 201 to 27,365 bp with an average length of 783 bp (Table 2). Further redundancy elimination using Evigene (Nakasugi et al. 2014) resulted in 69,373 unigenes with an average length of 1476 bp and N50 length of 2531 bp. In addition, there were 100,651 (38.3%) transcripts that exceeded 500 bp and 49,148 (18.7%) transcripts that exceeded 1000 bp. These results reflect the high quality of the de novo transcriptome assembly.
Functional annotation analysis of the 69,373 unigenes was carried out by using BLASTX with an E-value cutoff of 10−5. As a result, we annotated 34,011 (49.1%) unigenes against the NR database, 47,458 (68.4%) unigenes against NT database, and 27,221 (39.2%) unigenes against Swiss-prot database. Twenty-six thousand two hundred nineteen (37.8%) and 10,365 (14.9%) unigenes were annotated against GO and KEGG databases, respectively. All in all, 48,377 (69.7%) unigenes had at least one hit from these databases (Table 3).
Analysis of DEGs at Three Time Points after Challenge
In order to identify the DEGs between control and challenge groups at three time points post-infection, we assessed the expression level of each unigene in all 18 libraries. The gene expression profiles in control and challenge groups were compared at three time points. If a gene’s expression in challenge groups at corresponding time points exceeded a twofold difference relative to baseline (p ≤ 0.05), this gene was regarded as a DEG. As a result, a total of 1890 unigenes were found to be significantly differentially expressed in at least one time point post-infection. The FPKM value and annotation for DEGs are summarized in Supplementary Table 1 and Supplementary Table 2, respectively. In detail, 199, 1131, and 1140 unigenes were differentially expressed at 4, 24, and 48 h, respectively, after challenge relative to the control group. The number of DEGs at 24 h after challenge rose to 1131 due to a marked increase of downregulated and upregulated DEGs. The number of DEGs at 48 h after the challenge was 1140, which was roughly equivalent to the number of DEGs at 24 h (Table 4). The overall number of upregulated (1216) and downregulated (1254) DEGs were similar; however, at 4 h after infection, the number of upregulated DEGs (172) was more than six time as many as that of downregulated genes (27) (Table 4), indicating that numerous genes were activated even at the early stage of infection, although we failed to identify typical symptoms by eyes. There were 455 upregulated and 676 downregulated genes at 24 h post-infection, and 589 upregulated and 551 downregulated genes at 48 h post-infection.
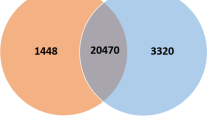
As shown in Fig. 2, 75 DEGs were shared in the comparisons in all three time points and are annotated in Supplementary Table 3. The number of exclusively shared DEGs between 4 and 24 h (16) and between 4 and 48 h (15) was obviously smaller than that between 24 and 48 h (399), reflecting the gene expression profile change during the infection stages. The annotation of the genes are shown in Supplementary Tables 4–6. Furthermore, we found 93, 641, and 651 unique DEGs for the 4-, 24-, and 48-h comparisons (Fig. 2).

Venn diagram showing commonalities and differences at three time points of shared differentially expressed genes
Identification of the Continuously Upregulated or Downregulated DEGs Post-Infection
If the FPKM value of a unigene at any time point was at least 1.5-fold of the FPKM value at the previous time point, this unigene during the A. hydrophila infection was identified as a continuously upregulated unigene. Using this criterion, we obtained 32 continuously upregulated unigenes during the three A. hydrophila infection stages (Supplementary Table 7). These include leptin-B, which is involved in food intake, fat metabolism, stress, and immune function (Copeland et al. 2011; Yuan et al. 2014a), implying the gene may be regulated during the infections. The expression of leptin-B at 4 h in the challenge groups was almost 29-fold of the expression at 4 h in the control groups. Its expressions at 24 and 48 h post-infection samples were almost 7- and 26-fold, respectively, of the expression at 4 h in the challenge groups. In addition, we also found that IL1R2 and CD59 glycoprotein, which play important roles in IL-1ß activity (Moltó and Olivé 2010) and complement inhibition (Zhang et al. 2015b), respectively, were continuously upregulated. The expressions of IL1R2 at 24 and 48 h post-infection samples were almost 11- and 167-fold, respectively, of the expression at 4 h in the challenge groups. The largest fold change was that of CD59 glycoprotein, whose expressions at 24 and 48 h in the challenge groups were almost 146- and 237-fold, respectively, of the expression at 4 h in the challenge groups.
Similarly, 12 continuously downregulated genes were also identified at all three time points, among which seven genes are annotated and summarized in Supplementary Table 7. Among these continuously downregulated genes, thioredoxin-interacting protein (TXNIP) is a physiological inhibitor of thioredoxin (Trx) expression and activity; its effects include direct activation of inflammation, apoptotic pathways, and increase in oxidative stress (Chong et al. 2014). The expressions of TXNIP at 24 and 48 h post-infection samples were almost 2- and 15-fold of that at 4 h post-infection samples.
Enrichment Analysis and Cluster Classification of DEGs
All the DEGs at each time point were analyzed according to GO functional enrichment analysis (Supplementary Table 8). The top three GO terms at 4 h post-infection involved in biological processes included single-organism process (GO:0044699), response to stimulus (GO:0050896), and single-multicellular organism process (GO:0044707). The top three molecular functions at 4 h post-infection identified in this analysis were protein binding (GO:0005515), receptor binding (GO:0005102), and peptidase activity (GO:0008233). The top three cellular components included extracellular region (GO:0005576), extracellular region part (GO:0044421), and membrane-bounded vesicle (GO:0031988).
Analysis of the Immune-Related DEGs During A. hydrophila Challenge
In the KEGG annotation, we identified 513 putative immune-related genes from the transcriptome, among which 59 genes were significantly differentially expressed (Supplementary Table 9). Immune-related DEGs were annotated to the immune system and 15 pathways (Supplementary Table 10), including complement and coagulation cascades (17 DEGs), chemokine signaling pathway (9 DEGs), platelet activation (8 DEGs), and toll-like receptor signaling pathway (7 DEGs). Nineteen, 36, and 30 immune-related DEGs were identified at 4, 24, and 48 h, respectively (Supplementary Table 9).
Validation of RNA-Seq Profiles by QPCR
To validate the gene profiles from RNA-seq, expression of randomly selected 15 genes related to immune system was measured by quantitative real-time PCR analyses (Fig. 3). As a result, the melting-curve analysis showed a single product for all selected genes. Gene expression from RNA-seq was significantly correlated with the QPCR results at each time point (average correlation coefficient of 0.84) with few exceptions, including genes CCL21, TLR3, MYD88, and HSPA1/8, showing different gene expression patterns for several time points. However, the overall consistency of expression from RNA-seq and QPCR for the majority of genes revealed the sequence assembly and the expression analysis of genes provided us a solid transcriptome information in this work.

Comparison of relative fold change between QRT-PCR and RNA-seq results in S. prenanti spleen at (A) 4 h, (B) 24 h, and (C) 48 h. Gene abbreviations are the following: CCL21, C-C motif chemokine 21; C1QA, complement C1q subcomponent subunit A; HSPA1-8, heat shock 70 kDa protein 1/8; IL6R, interleukin 6 receptor; TLR3, toll-like receptor 3; TLR4, toll-like receptor 4; TLR5, toll-like receptor 5; TNFAIP3, tumor necrosis factor, alpha-induced protein 3; MYD88, myeloid differentiation primary response protein MyD88; TOLLIP, toll-interacting protein; TRAF6, TNF receptor-associated factor 6; MHC1, major histocompatibility complex, class I; IL1R2, interleukin 1 receptor type II; IL12A, interleukin 12A; ITGA5, integrin alpha 5. A, B, and C represent 4, 24, and 48 h time points, respectively. Fold changes are expressed as the ratio of gene expression after A. hydrophila challenge to the control group as normalized with β-actin. Asterisk indicates significant (p < 0.05) fold at a given time point relative to control
Discussion
We assembled and analyzed the spleen transcriptomes of S. prenanti at 4, 24, and 48 h after the injection of A. hydrophila. As far as we know, this is the first large-scale investigation of time-series gene expression patterns with respect to S. prenanti-A. hydrophila interaction. The reliability of the DGE results obtained from RNA-seq was conformed by QPCR. The gene expression patterns and relative expression levels of vast majority selected genes were consistent. This result resembled those of Misgurnus anguillicaudatus (Huang et al. 2016), Ctenopharyngodon idella (Song et al. 2017b), and C. macrocephalus (Chatchaiphan et al. 2017).
After analysis of spleen transcriptome of S. prenanti at 4, 24, and 48 h after the challenge experiments, 199, 1131, and 1140 DEGs (totally 1890) were identified, respectively. This is similar to the results obtained at three time points within 3–72 h after artificial challenge in the transcriptome studies for Ictalurus Punetaus (Li et al. 2012; Sun et al. 2012). Obviously, the number of DEGs in spleen sharply increased at 0–24 h, while the DEG number at 24–48 h is relatively stable. We speculated that at the initial 4 h after A. hydrophila invasion of S. prenanti, bacterial proliferation was slow and the degree of stimulation of fish immune system was relatively mild. One possible reason to explain the result could be that the immune systems of S. prenanti were fully activated to react to the invasion after 24 and 48 h post-infection; therefore, the number of DEGs in 24 and 48 h was comparable. However, we noticed that only 1/3 DEGs were shared between 24 and 48 h, suggesting different immune reactions were probably carried out. As we expected, numerous DEGs were annotated in the defense systems of S. prenanti at the three time points, such as response to stimulus (871 DEGs), cellular response to stimulus (585 DEGs), defense response (151 DEGs), immune system process (142 DEGs), response to bacterium (88 DEGs), and defense response to bacterium (40 DEGs), which was consistent with the gene expression study of other fishes after bacterial infection (Zhang et al. 2015b; Zhou et al. 2015; Zhu et al. 2015).
KEGG is a database to understand the advanced functions of biological systems and utilizes biological systems at the molecular level. In this study, 1890 DEGs of three time points were respectively enriched in 27 (4 h, 105 unigenes), 36 (24 h, 289 unigenes), and 44 (48 h, 349 unigenes) KEGG signaling pathways. Most genes of the three time points, especially at 4 h, were significantly enriched in the upstream and downstream signaling pathways associated with immunity. This is similar to the research results for I. Punetaus (Li et al. 2012; Beck et al. 2012), Danio rerio (Lü et al. 2015), M. amblycephala (Tran et al. 2015), and Oreochromis niloticus (Zhang et al. 2013). Further analysis found that 59 significantly differentially expressed genes were involved in immune system signaling pathways and the range of expression fold change was − 4.32~11.16.
By comparing infected samples to the controls for each time point, many immune-related genes were identified, and we believed that those genes played an important role of A. hydrophila invasion reactions for S. prenanti, since they have already been well investigated for their contribution to the immune reaction upon pathogen infections in previous researches. Contrasted with the control group, 18 immune-related genes were significantly differentially expressed and all of those genes were activated at 4 h. Claudin genes were upregulated by 8.14 times at 4 h compared with the control group. Claudins are the main components of cell tight junction molecules and the main determining molecules of the paracellular shunt pathway, and mediate intercellular material transport through size selection and charge selection (Aijaz et al. 2006). The abnormal expression of claudin genes could lead to a structural and functional destruction of endothelial cells and epithelial cells and thus is closely related to the occurrence and development of many diseases. We speculated that S. prenanti upregulated the expression of claudin genes to strengthen the tight junctions between cells, especially in epithelial tissues, so as to prevent the invasion of A. hydrophila in the early stages of infection. Thirty-six immune-related genes were significantly differentially expressed at 24 h. Thirteen of them were significantly downregulated and 23 were significantly upregulated. The most downregulated is natural cytotoxicity triggering receptor 1 (NCR1, FC = − 4.32) and the most upregulated is interleukin 12A (IL12A, FC = +11.16). NCR1 is one of the major triggering receptors of natural killer cells that could induce mobilization of calcium and cytotoxicity and cause cytokines to be released. It could also serve as a receptor to participate in the dissolution of many targets, while IL12 is a proinflammatory cytokine with multiple immune regulation functions and an NK cell activating factor with the strongest effect. IL12 could not only activate NK precursor cells and stimulate proliferation and activation of NK cells but also play an important role in acquired immune responses. Twenty-eight immune-related genes were significantly differentially expressed at 48 h, among which six were significantly downregulated and 22 were significantly upregulated. The most downregulated is alpha-1-antitrypsin (A1AT, FC = − 3.18) and the most upregulated is interleukin 11 (IL11, FC = + 11.03). A1AT is a protease inhibitor that can inhibit neutrophil elastase, thrombin, plasma endothelin, and other serine endopeptidases. It plays an important role in protecting the body of normal cells and organs from protease damage, blocking or delaying bacterial growth, inhibiting inflammation and infection, and maintaining the stability of the internal environment (Zhuang et al. 2007; Magnadottir 2010). IL11 is a member of the glycoprotein-130 (GP-130) cytokine family and it is traditionally considered being an anti-inflammatory cytokine. However, recent research showed that it is also a proinflammatory cytokine under certain conditions and it has different immune regulatory functions in different cells (Xu et al. 2016).
Meanwhile, we also found that eight immune-related genes were upregulated significantly through all infection stages, including matrix metalloproteinase-9 (gelatinase B, MMP9), interleukin 11 (IL11), urokinase plasminogen activator (PLAU), neutrophil cytosolic factor 1 (NCF1), interleukin 1 beta (IL1B), tumor necrosis factor, alpha-induced protein 3 (TNFAIP3), coagulation factor III (tissue factor,CD142, or F3), and interleukin 1 receptor type 2 (IL1R2). Eleven immune-related genes were upregulated significantly, and only one was downregulated (guanylate cyclase soluble subunit beta, GUCY1B) at 24 and 48 h. MMP9 is a matrix metalloproteinase (MMP) multigene family member, which plays a vital role in cell migration and tissue remodeling during inflammation in teleosts (Xu et al. 2013). Hence, MMP9 might play a key role in both initiation and termination of the inflammatory processes (Chadzinska et al. 2008). The previous studies reported that the expression of mmp9 can be affected by A. hydrophila infection (Xu et al. 2013; Jiang et al. 2016; Yang et al. 2016; Song et al. 2017b), indicating that MMP9 plays an important role in A. hydrophila-related diseases. IL1R2 was the first decoy receptor for the IL-1 system to be depicted, which binds IL-1α and IL-1β and acts as a molecular trap for the agonists of the signaling IL1R1 to negatively regulate the IL1 signaling in the inflammatory response (Zhu et al. 2013; Bonecchi et al. 2016). In addition, IL1R2 exerts an anti-inflammatory role in several diseases (Rauschmayr et al. 1997; Bonecchi et al. 2016). In this study, IL1R2 expression was rapidly upregulated after being challenged with A. hydrophila, and reached the highest expression level at 48 h, which is similar with the previous report in common carp (Jiang et al. 2016).
Conclusion
In summary, we have investigated S. prenanti transcriptomic response in the spleen after A. hydrophila challenge applying RNA-seq technology for the first time. Among 69,373 assembled and functionally annotated unigenes, 1890 DEGs were identified in S. prenanti spleen at 4, 24, and 48 h after the artificial challenge, including 59 immune-related genes. We revealed a time series of gene expression profiles of the immune system in S. prenanti upon A. hydrophila infection and found that gene expression patterns and the functional enrichment of DEGs reflected the stepwise molecular responses to the invasion of A. hydrophila at different infection stages. Based on our results, many important functional genes involved in the immune system, such as claudin, ncr1, atat, il12, and il1r2, were found to play potential key roles in reacting against A. hydrophila infection; however, their biological functions need further experimental investigation in our following studies. The results would advance our understanding of the gene regulation and immune response at the different stages of the infection of A. hydrophila in S. prenanti and provide useful information to take appropriate measures to control and to prevent the disease.
References
Aijaz S, Balda MS, Matter K (2006) Tight junctions: molecular architecture and function. Int Rev Cytol 248:261–298
Altschul S, Madden T, Schaffer A et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Beck BH, Farmer BD, Straus DL, Li C, Peatman E (2012) Putative roles for a rhamnose binding lectin in Flavobacterium columnare pathogenesis in channel catfish Ictalurus punctatus. Fish Shellfish Immunol 33:1008–1015
Bonecchi R, Garlanda C, Mantovani A, Riva F (2016) Cytokine decoy and scavenger receptors as key regulators of immunity and inflammation. Cytokine 87:37–45
Chadzinska M, Baginski P, Kolaczkowska E, Savelkoul HFJ, Lidy Verburg-van Kemenade BM (2008) Expression profiles of matrix metalloproteinase 9 in teleost fish provide evidence for its active role in initiation and resolution of inflammation. Immunology 125:601–610
Chatchaiphan S, Srisapoome P, Kim J-H, Devlin RH, Na-Nakorn U (2017) De novo transcriptome characterization and growth-related gene expression profiling of diploid and triploid bighead catfish (Clarias macrocephalus Günther, 1864). Mar Biotechnol 19:36–48
Chong C-R, Chan WPA, Nguyen TH, Liu S, Procter NEK, Ngo DT, Sverdlov AL, Chirkov YY, Horowitz JD (2014) Thioredoxin-interacting protein: pathophysiology and emerging pharmacotherapeutics in cardiovascular disease and diabetes. Cardiovasc Drugs Ther 28:347–360
Colwell R, Macdonell M, Deley J (1986) Proposal to recognize the family Aeromonadaceae fam-Nov. Int J Syst Bacteriol 36:473–477
Conesa A, Götz S, García-Gómez JM et al (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Copeland DL, Duff RJ, Liu Q, Prokop J, Londraville RL (2011) Leptin in teleost fishes: an argument for comparative study. Front Physiol 2:26
Ding R (1994) The fishes of Sichuan (Chinese). Sichuan Publishing House of Science and Technology, Chengdu, Sichuan, China
Du X, Li Y, Li D et al (2017) Transcriptome profiling of spleen provides insights into the antiviral mechanism in Schizothorax prenanti after poly (I: C) challenge. Fish Shellfish Immunol 62:13–23
Du Z, Huang X, Wang K et al (2011) Isolation and identification of etiology of skin ulcer in Schizothorax prenanti (Chinese). J Sichuan Agric Univ:274–279
Du Z, Wang K, Li Z, Fan L (2003) The first reports of disease control and prevention in Schizothorax prenanti (Chinese). China Fish 82–83
Geng Y, Wang KY, Huang XL, Chen DF, Li CW, Ren SY, Liao YT, Zhou ZY, Liu QF, du ZJ, Lai WM (2012) Streptococcus agalactiae, an emerging pathogen for cultured Ya-fish, Schizothorax prenanti, in China. Transbound Emerg Dis 59:369–375
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Huang S, Cao X, Tian X (2016) Transcriptomic analysis of compromise between air-breathing and nutrient uptake of posterior intestine in loach (Misgurnus anguillicaudatus), an air-breathing fish. Mar Biotechnol 18:521–533
Janda JM, Abbott SL (2010) The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin Microbiol Rev 23:35–73
Jiang Y, Feng S, Zhang S, Liu H, Feng J, Mu X, Sun X, Xu P (2016) Transcriptome signatures in common carp spleen in response to Aeromonas hydrophila infection. Fish Shellfish Immunol 57:41–48
Joseph SW, Carnahan A (1994) The isolation, identification, and systematics of the motile Aeromonas species. Annu Rev Fish Dis 4:315–343
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323
Li C, Zhang Y, Wang R, Lu J, Nandi S, Mohanty S, Terhune J, Liu Z, Peatman E (2012) RNA-seq analysis of mucosal immune responses reveals signatures of intestinal barrier disruption and pathogen entry following Edwardsiella ictaluri infection in channel catfish, Ictalurus punctatus. Fish Shellfish Immunol 32:816–827
Li Y, Lai S, Wang R, Zhao Y, Qin H, Jiang L, Li N, Fu Q, Li C (2017) RNA-Seq analysis of the antioxidant status and immune response of Portunus trituberculatus following aerial exposure. Mar Biotechnol 19:89–101
Liang J, Liu Y, Zhang X, Zhang X, Yue B, Song Z (2011) An observation of the loss of genetic variability in prenant’s schizothoracin, Schizothorax prenanti, inhabiting a plateau lake. Biochem Syst Ecol 39:361–370
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408
Lü A-J, Hu X-C, Wang Y, Zhu AH, Shen LL, Tian J, Feng ZZ, Feng ZJ (2015) Skin immune response in the zebrafish, Danio rerio (Hamilton), to Aeromonas hydrophila infection: a transcriptional profiling approach. J Fish Dis 38:137–150
Luo H, Xiao S, Ye H, Zhang Z, Lv C, Zheng S, Wang Z, Wang X (2016) Identification of immune-related genes and development of SSR/SNP markers from the spleen transcriptome of Schizothorax prenanti. PLoS One 11:e0152572
Magnadottir B (2010) Immunological control of fish diseases. Mar Biotechnol 12:361–379
Moltó A, Olivé A (2010) Anti-IL-1 molecules: new comers and new indications. Joint Bone Spine 77:102–107
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Mu Y, Ding F, Cui P, Ao J, Hu S, Chen X (2010) Transcriptome and expression profiling analysis revealed changes of multiple signaling pathways involved in immunity in the large yellow croaker during Aeromonas hydrophila infection. BMC Genomics 11:506
Nakasugi K, Crowhurst R, Bally J, Waterhouse P (2014) Combining transcriptome assemblies from multiple de novo assemblers in the allo-tetraploid plant Nicotiana benthamiana. PLoS One 9:e91776
Rauschmayr T, Groves RW, Kupper TS (1997) Keratinocyte expression of the type 2 interleukin 1 receptor mediates local and specific inhibition of interleukin 1-mediated inflammation. Proc Natl Acad Sci U S A 94:5814–5819
Robinson N, Sahoo PK, Baranski M, Mahapatra KD, Saha JN, Das S, Mishra Y, Das P, Barman HK, Eknath AE (2012) Expressed sequences and polymorphisms in Rohu carp (Labeo rohita, Hamilton) revealed by mRNA-seq. Mar Biotechnol 14:620–633
Singh V, Chaudhary DK, Mani I, Jain R, Mishra BN (2013) Development of diagnostic and vaccine markers through cloning, expression, and regulation of putative virulence-protein-encoding genes of Aeromonas hydrophila. J Microbiol (Seoul, Korea) 51:275–282
Song J, Song Z, Yue B, Zheng W (2006) Assessing genetic diversity of wild populations of Prenant’s schizothoracin, Schizothorax prenanti, using AFLP markers. Environ Biol Fish 77:79–86
Song K, Li L, Zhang G (2017a) Bias and correction in RNA-seq data for marine species. Mar Biotechnol 19:541–550
Song X, Hu X, Sun B, Bo Y, Wu K, Xiao L, Gong C (2017b) A transcriptome analysis focusing on inflammation-related genes of grass carp intestines following infection with Aeromonas hydrophila. Sci Rep 7:40777
Sun F, Peatman E, Li C, Liu S, Jiang Y, Zhou Z, Liu Z (2012) Transcriptomic signatures of attachment, NF-κB suppression and IFN stimulation in the catfish gill following columnaris bacterial infection. Dev Comp Immunol 38:169–180
Tran NT, Gao Z-X, Zhao H-H, Yi SK, Chen BX, Zhao YH, Lin L, Liu XQ, Wang WM (2015) Transcriptome analysis and microsatellite discovery in the blunt snout bream (Megalobrama amblycephala) after challenge with Aeromonas hydrophila. Fish Shellfish Immunol 45:72–82
Wang L, Feng Z, Wang X, Wang X, Zhang X (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138
Wei R, Liu T, Zhou C, Zhang XD, Yuan DY, Wang T, Lin FJ, Chen H, Wu HW, Li ZQ (2013a) Identification, tissue distribution and regulation of preproghrelin in the brain and gut of Schizothorax prenanti. Regul Pept 186:18–25
Wei R, Yuan D, Wang T, Zhou CW, Lin FJ, Chen H, Wu HW, Yang SY, Wang Y, Liu J, Gao YD, Li ZQ (2013b) Characterization, tissue distribution and regulation of agouti-related protein (AgRP) in a cyprinid fish (Schizothorax prenanti). Gene 527:193–200
Wei R, Yuan D, Zhou C, Wang T, Lin F, Chen H, Wu H, Xin Z, Yang S, Chen D, Wang Y, Liu J, Gao Y, Li Z (2013c) Cloning, distribution and effects of fasting status of melanocortin 4 receptor (MC4R) in Schizothorax prenanti. Gene 532:100–107
Xu DH, Zhu Z, Wakefield MR, Xiao H, Bai Q, Fang Y (2016) The role of IL-11 in immunity and cancer. Cancer Lett 373:156–163
Xu X-Y, Shen Y-B, Fu J-J, Liu F, Guo SZ, Li JL (2013) Characterization of MMP-9 gene from grass carp (Ctenopharyngodon idella): an Aeromonas hydrophila-inducible factor in grass carp immune system. Fish Shellfish Immunol 35:801–807
Yang X, Liu D, Liu F, Wu J, Zou J, Xiao X, Zhao F, Zhu B (2013) HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinformatics 14:33
Yang Y, Yu H, Li H, Wang A (2016) Transcriptome profiling of grass carp (Ctenopharyngodon idellus) infected with Aeromonas hydrophila. Fish Shellfish Immunol 51:329–336
Ye H, He L, Cheng H et al (2014) Changes of cell immunity indexes in peripheral blood of Schizothorax prenanti injected with formalin-killed Aeromonas hydrophila. J Southwest Univ National (Nat Sci Ed) 36:28–35
Yuan D, Wang T, Zhou C, Lin F, Chen H, Wu H, Wei R, Xin Z, Li Z (2014a) Leptin and cholecystokinin in Schizothorax prenanti: molecular cloning, tissue expression, and mRNA expression responses to periprandial changes and fasting. Gen Comp Endocrinol 204:13–24
Yuan D, Zhou C, Wang T, Lin F, Chen H, Wu H, Wei R, Xin Z, Liu J, Gao Y, Chen D, Yang S, Wang Y, Pu Y, Li Z (2014b) Molecular characterization and tissue expression of peptide YY in Schizothorax prenanti: effects of periprandial changes and fasting on expression in the hypothalamus. Regul Pept 190–191:32–38
Zhang R, Zhang L, Ye X, Tian YY, Sun CF, Lu MX, Bai JJ (2013) Transcriptome profiling and digital gene expression analysis of Nile tilapia (Oreochromis niloticus) infected by Streptococcus agalactiae. Mol Biol Rep 40:5657–5668
Zhang X, Gao X, Wang J, Cao W (2015a) Extinction risk and conservation priority analyses for 64 endemic fishes in the upper Yangtze River, China. Environ Biol Fish 98:261–272
Zhang X, Wang S, Chen S, Chen Y, Liu Y, Shao C, Wang Q, Lu Y, Gong G, Ding S, Sha Z (2015b) Transcriptome analysis revealed changes of multiple genes involved in immunity in Cynoglossus semilaevis during Vibrio anguillarum infection. Fish Shellfish Immunol 43:209–218
Zhou W, Zhang Y, Wen Y, Ji W, Zhou Y, Ji Y, Liu X, Wang W, Asim M, Liang X, Ai T, Lin L (2015) Analysis of the transcriptomic profilings of mandarin fish (Siniperca chuatsi) infected with Flavobacterium columnare with an emphasis on immune responses. Fish Shellfish Immunol 43:111–119
Zhu J, Li C, Ao Q, Tan Y, Luo Y, Guo Y, Lan G, Jiang H, Gan X (2015) Trancriptomic profiling revealed the signatures of acute immune response in tilapia (Oreochromis niloticus) following Streptococcus iniae challenge. Fish Shellfish Immunol 46:346–353
Zhu L, Nie L, Zhu G, Xiang LX, Shao JZ (2013) Advances in research of fish immune-relevant genes: a comparative overview of innate and adaptive immunity in teleosts. Dev Comp Immunol 39:39–62
Zhu Q, Zhang L, Li L, Que H, Zhang G (2016) Expression characterization of stress genes under high and low temperature stresses in the Pacific oyster, Crassostrea gigas. Mar Biotechnol 18:176–188
Zhuang M, Ai Z, Wu M (2007) Development of alpha-1-antitrypsin. J Microbiol 27:92–95
Acknowledgments
We thank Lingbing Zeng, in Yangtze River Fisheries Research Institute Chinese Academy of Fishery Sciences, and Bin Chen, in College of Animal Science and Technology, Hunan Agricultural University, for providing constructive suggestions on the experiments. We also thank Genhua Yue and Baoqing Ye, Molecular Population Genetics Group, Temasek Life Sciences Laboratory, 1 research link, National University of Singapore, for providing constructive suggestions on the manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (31402302, 31602207), Fundamental Research Funds for the central Universities (XDJK2015C034, XDJK2017B008, XDJK2017C035), Scientific Research Initiation Project aided by a special fund, Southwest University Rongchang Campus (20700208), and the Youth Foundation of Southwest University Rongchang Campus (20700937, 20700938).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Table 1
(XLSX 145 kb)
Supplementary Table 2
(XLSX 139 kb)
Supplementary Table 3
(XLSX 14 kb)
Supplementary Table 4
(XLSX 9 kb)
Supplementary Table 5
(XLSX 9 kb)
Supplementary Table 6
(XLSX 38 kb)
Supplementary Table 7
(XLSX 15 kb)
Supplementary Table 8
(XLSX 16 kb)
Supplementary Table 9
(XLSX 17 kb)
Supplementary Table 10
(XLSX 14 kb)
Rights and permissions
About this article

Cite this article
Ye, H., Xiao, S., Wang, X. et al. Characterization of Spleen Transcriptome of Schizothorax prenanti during Aeromonas hydrophila Infection. Mar Biotechnol 20, 246–256 (2018). https://doi.org/10.1007/s10126-018-9801-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-018-9801-0