
Abstract
Long noncoding RNAs (lncRNAs) have been recognized in recent years as key regulators of diverse cellular processes. Genome-wide large-scale projects have uncovered thousands of lncRNAs in many model organisms. Large intergenic noncoding RNAs (lincRNAs) are lncRNAs that are transcribed from intergenic regions of genomes. To date, no lincRNAs in non-model teleost fish have been reported. In this report, we present the first reference catalog of 9674 rainbow trout lincRNAs based on analysis of RNA-Seq data from 15 tissues. Systematic analysis revealed that lincRNAs in rainbow trout share many characteristics with those in other mammalian species. They are shorter and lower in exon number and expression level compared with protein-coding genes. They show tissue-specific expression pattern and are typically co-expressed with their neighboring genes. Co-expression network analysis suggested that many lincRNAs are associated with immune response, muscle differentiation, and neural development. The study provides an opportunity for future experimental and computational studies to uncover the functions of lincRNAs in rainbow trout.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Long noncoding RNAs (lncRNAs) are RNA molecules that are longer than 200 nucleotides and carry many signatures of mRNAs, such as 5′ capping, 3′ polyadenylation, and RNA splicing, but have little or no open reading frame (Bhartiya et al. 2012; Liao et al. 2011; Carninci et al. 2005). They have emerged as a new class of regulatory transcripts in recent years (Perkel 2013; Khaitovich et al. 2006). Recent advances in sequencing technologies have opened a new horizon for the identification and annotation of this class of RNAs in many species. The lncRNAs that are transcribed from intergenic regions of genomes are termed large intergenic noncoding RNAs (lincRNAs). As lincRNAs do not overlap with protein-coding regions, it makes computational analysis of such RNAs easier. To date, at least 15,512 human lincRNAs and over 10,000 mouse lincRNAs have been identified (Derrien et al. 2012; Luo et al. 2013).
Recent studies have supported the view that lincRNAs play important roles in many biological processes, such as procession of p53 response pathways (Huarte et al. 2010; Loewer et al. 2010; Hung et al. 2011), regulation of epigenetic marks and gene expression (Rinn et al. 2007; Zhao et al. 2008; Khalil et al. 2009; Pandey et al. 2008), maintenance of pluripotency (Guttman et al. 2009), and activation of gene expression as “enhancer RNAs” (Orom et al. 2010; Wang et al. 2011). In addition, lincRNAs have also been associated with human diseases and pathophysiological conditions (Gupta et al. 2010; Zhu et al. 2011; Cabianca et al. 2012).
Rainbow trout (Oncorhynchus mykiss) is a species of salmonid native to cold-water tributaries of the Pacific Ocean in Asia and North America. It is one of the most important cold-water fish species in the USA due to its importance for food production, sport fisheries, and as a research model (Thorgaard et al. 2002). To generate genomic resources for genetic studies of this species, we have characterized the rainbow trout mRNA and microRNA transcriptomes (Ma et al. 2012; Salem et al. 2010a, b, 2015). In particular, a complete transcriptome has been generated by RNA sequencing of cDNA libraries from multiple tissues of a single-doubled haploid rainbow trout (Salem et al. 2015). With the increasing evidence supporting important roles of lincRNAs in diverse processes, a systematic catalog of these RNA transcripts and their expression across tissues in rainbow trout is warranted. The recent publication of rainbow trout genome sequence (Berthelot et al. 2014) and computational methods for transcriptome reconstruction (Guttman et al. 2010; Trapnell et al. 2009; Garber et al. 2011) provide an opportunity to comprehensively annotate and characterize lincRNA transcripts in rainbow trout.
Here we report the systematic identification and characterization of lincRNAs in 15 major tissue types of rainbow trout. We analyzed the known genomic features of the identified lincRNAs including transcript length, exon number, and spatiotemporal expression specificity. We also used weighted gene co-expression network to assign functionalities to the lincRNAs, which revealed that lincRNAs are expressed in a strong tissue-specific manner, and many of them are highly associated with biological processes specific to that tissue (e.g., a brain-specific group is enriched with functional terms such as neural development and axon injury response). This study is the first report of a genome-wide annotation of rainbow trout lincRNAs, which will facilitate future experimental and computational studies to uncover the functions of lincRNAs in rainbow trout.
Materials and Methods
Tissue Sample Collection and RNA Sequencing
Tissue collection and RNA sequencing were described in detail in a previous study (Salem et al. 2015). In brief, 13 different tissues were collected from a single male homozygous rainbow trout, which was euthanized under protocol no. 02456 approved by the Washington State University Institutional Animal Care and Use Committee. These tissues include the brain, fat, gill, head kidney, intestine, kidney, liver, testis, red muscle, skin, spleen, stomach, and white muscle. In addition, oocyte and pineal samples were collected from different fish. Total RNA from each sample was isolated using Trizol (Invitrogen, Carlsbad, CA). Library construction and sequencing were performed at Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. Each library was loaded onto one lane and paired-end sequencing with 2 × 100 cycles was performed on an Illumina Genome Analyzer IIx (Illumina, San Diego, CA).
RNA-Seq Reads Mapping and Transcriptome Assembly
Spliced read aligner TopHat version V2.0 (Trapnell et al. 2009) was used to map all sequence reads to the rainbow trout genome (Berthelot et al. 2014). A two-step mapping process was performed by TopHat using the following parameters: min-anchor = 5, min-isoform-fraction = 0, and default values for the remaining parameters. Bowtie2 (Langmead and Salzberg 2012) was used first to align reads with no gaps that can directly map to the genome reference sequence. Gapped alignment was then performed to align the reads that were not aligned in the first step. The aligned reads from each sample were assembled into transcriptome by Cufflinks version V2.2.1 (Trapnell et al. 2010) that uses spliced reads information to determine exons connectivity. The Cufflinks assembler generates the output in the form of fragments per kilo base of exons per million fragments generated (FPKM) value, which is directly proportional to the relative abundance of a transcript in a given sample.
FPKM Threshold for Classifying Complete and Partial Transcripts
Individual transcript assembly may have noise from multiple sources such as artifacts generated by sequence alignment, unspliced intronic pre-mRNA, or genomic DNA contamination. Sebnif (Sun et al. 2014), an integrative bioinformatics pipeline that identifies high-quality single- and multi-exonic lincRNAs by optimizing a FPKM threshold, was used to minimize the assembly noise and enhance the quality of identified lincRNAs. Considering the difference of the structure between the multi- and single-exonic transcripts, two separate algorithms were used to identify the optimal FPKM thresholds. (1) For multi-exonic transcripts, a fully reconstruction fraction estimation (FRFE) approach was used by Sebnif (Guttman et al. 2010). Briefly, multi-exonic transcripts in reference annotation were first divided into N expression quantiles based on their FPKM values. At each expression quantile, the reference transcript set was then divided into two categories, fully reconstructed transcripts and partially reconstructed transcripts. The assembly quality was evaluated by the proportion of the fully reconstructed transcripts, which is also called fully reconstruction fraction (FRF), at each expression quantile. The index of the optimum FPKM threshold was obtained by balancing the sensitivity and specificity based on the FRF value with the following formula (Sun et al. 2012):
where i* is the index of FPKM threshold for each quantile i. The sensitivity [i] and specificity [i] indicate the ith sensitivities and specificities, respectively. The i belongs to [1, N]. The optimum FPKM threshold was generated by pROC (Robin et al. 2011). (2) For single-exonic transcripts, single-exonic transcript Gaussian/gamma estimation (STGE) was implemented to estimate the optimal expression threshold (Sun et al. 2014). In the STGE algorithm, the appropriate model was determined by fitting the expression values of the single-exonic transcripts in the reference annotation. Any transcript whose expression falls into either tail of fitting model distribution was considered unreliable and discarded.
LincRNA Detection Pipeline
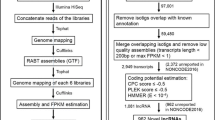
A step-wise filtering pipeline (Fig. 1) was used to identify putative lincRNAs from deep sequencing data. (1) All transcripts smaller than 200 bases were excluded. (2) Assembled transcripts were annotated using Cuffcompare from Cufflinks (Trapnell et al. 2010). Transcripts that are located in the intergenic region, at least 1 kb from any known protein-coding genes, were selected as putative lincRNAs (Luo et al. 2013). (3) The coding potential of each transcript was calculated using Coding-Potential Assessment Tool (CPAT) (Wang et al. 2013a) and Coding Potential Calculator (CPC) (Kong et al. 2007). (4) To evaluate which of the remaining transcripts contains a known protein-coding domain, HMMER-3 (Finn et al. 2011) was used to identify transcripts translated in all six possible frames having homologs with any of the 31,912 known protein family domains in the Pfam database (release 24; both PfamA and PfamB). All transcripts with a Pfam hit were excluded. (5) Putative protein-coding RNAs were filtered out by applying a maximal open reading frame (ORF) length threshold. Any transcripts with a maximal ORF > 100 amino acids was excluded. (6) Sequence homology search was performed to remove those transcripts with significant similarity with RNAs in several different public RNA databases including Rfam (Gardner et al. 2009), RNAdb (Pang et al. 2007), and lncRNAdb (Amaral et al. 2011). (7) The remaining transcripts that are at least 1 kb from any known protein-coding genes were selected (Luo et al. 2013).

Pipeline used to identify rainbow trout lincRNAs. a Raw RNA-Seq data was pre-processed and mapped using TopHat and assembled using Cufflinks in ab initio mode. b Sebnif was used to filter all lowly expressed unreliable transcripts. c Pipeline for lincRNA detection
Tissue Specificity Score and Neighboring Gene Correlation Analysis
To evaluate tissue specificity of a transcript, an entropy-based metric that relies on Jensen-Shannon (JS) divergence was used to calculate specificity scores (0 to 1). A perfect tissue-specific pattern is scored as JS = 1, which means a transcript is expressed only in one tissue (Cabili et al. 2011). In neighboring gene analysis, two genes were defined as neighbors if the minimal distance between them is <10 kb (regardless of their directions) (Zhang et al. 2014; Luo et al. 2013). The expression correlation between two neighbors was estimated by calculating the Pearson correlation coefficient between their density-normalized expression values (log2 FPKM + 1).
Weighted Gene Co-expression Network Construction and Gene Module Detection
All genes with expression variance ranked in the top 75 percentile of the data set were retained (Liao et al. 2011). R package “WGCNA” was then used to construct the weighted gene co-expression network (Langfelder and Horvath 2008). A matrix of signed Pearson correlation between all gene pairs was computed, and the transformed matrix (TOM) was used as input for linkage hierarchical clustering (Langfelder and Horvath 2008). Genes with similar expression patterns were clustered together.
Functional Enrichment Analysis
To investigate the potential roles of lincRNAs in rainbow trout, we performed Blast2GO (Conesa and Gotz 2008) analysis to assign gene ontology (GO) terms to all protein-coding genes associated with lincRNAs in each network module. A cutoff value of 1E−10 was used for the BLASTx search. GO term enrichment analysis was performed using Fisher’s exact test (p value <0.01). The interaction networks among lincRNA and protein-coding genes were constructed based on co-expression using Cytoscape (http://www.cytoscape.org/).
Validation of Expression Specificity of lincRNAs
Expression specificity of selected lincRNAs was validated by reverse transcription polymerase chain reaction (RT-PCR) analysis as described previously (Wang et al. 2013b). PCR primers are listed in Supplemental file 1. Tissue samples used in the analysis include the brain, fat, gill, head kidney, intestine, kidney, liver, testis, red muscle, skin, spleen, stomach, white muscle, oocyte, and pineal. 18S rRNA was used as a control for RNA quality.
Results and Discussion
Transcriptome Reconstruction and Filtering Low-Quality Assemblies
To comprehensively identify rainbow trout lincRNAs, we collected and deeply sequenced the RNA samples from the brain, fat, gill, head kidney, intestine, kidney, liver, testis, red muscle, skin, spleen, stomach, white muscle, oocyte, and pineal. A total of 1.3 billion raw paired-end sequence reads (100-bp read length) were generated from these samples. The number of reads from each tissue ranged from 78.8 to 93.5 million. A total of 1,087,497,866 cleaned reads (81.4 %) were harvested for further analysis. These sequence reads were mapped to the rainbow trout genome using TopHat (Trapnell et al. 2009), and approximately 447 million (82 %) mapped reads were recovered. The mapping ratio ranged from 76.9 to 89.5 % with an average of 82.3 % (Table 1). We then used the ab initio assemble software Cufflinks (Trapnell et al. 2010) to reconstruct the transcriptome for each tissue based on the read-mapping results (Fig. 1a). On average, 79,021 transcripts for each tissue were obtained.
The first challenge to annotate lincRNA gene loci is to distinguish lowly expressed lincRNAs from the tens of thousands of lowly expressed unreliable fragments assembled from RNA-Seq (Guttman et al. 2010). To address this challenge, we removed unreliable lowly expressed transcripts using a learned FPKM threshold, which was calculated using Sebnif (Sun et al. 2014) (Fig. 1b). First, we classified all transcripts that did not overlap the genomic region of known protein-coding genes as novel intergenic transcripts (category of “u” assigned by Cuffcompare) and defined an average of 28,012 u transcripts for each tissue (Fig. 1b; Supplemental file 2), among which 6975 and 21,037 are multi- and single-exonic transcripts, respectively. Next, FRFE and STGE algorithms were used to distinguish partial transcripts from full length transcripts. For 6975 multi-exonic transcripts, Sebnif applied a FRFE threshold of 0.5. For 21,037 single-exonic transcripts, STGE was used to model the transcript expression profiles with the lower and upper probability cutoffs set at 0.05 and 0.95, respectively. Following this filtering, an average of 4628 multi-exonic (FPKM >2.76) and 4071 single-exonic (FPKM >3.14) transcripts for each tissue were retained. Finally, a total of 39,745 intergenic transcripts were obtained by merging all intergenic transcripts from 15 tissues.
Identification and Characterization of Rainbow Trout lincRNAs
The currently available coding potential prediction methods only work well for protein-coding RNAs. Therefore, the most widely used strategy to annotate potential noncoding RNAs (ncRNA) is to exclude those that possess protein-coding features (Solda et al. 2009). The filtering pipeline we used to identify novel lincRNAs is shown in Fig. 1c. First, we analyzed the coding potential of unannotated transcripts using CPAT (Wang et al. 2013a) and CPC (Kong et al. 2007), which filtered out 61 % (24,329) of all transcripts. Second, we scanned each transcript in all six frames to exclude transcripts that contain any of the 31,912 protein-coding domains cataloged in the protein family database Pfam (Finn et al. 2008). This filtering retained 10,773 potential lincRNA transcripts. Furthermore, a minimal ORF length criterion was applied to distinguish lincRNAs from mRNAs. A cutoff of 300 nt (100 codons) was used to exclude putative mRNAs (Okazaki et al. 2002). For the characterization of ncRNAs not yet annotated in the rainbow trout genome assembly, sequence homology search was performed to exclude those transcripts with significant similarity with RNAs in Rfam (Gardner et al. 2009), RNAdb (Pang et al. 2007), and lncRNAdb (Amaral et al. 2011). Finally, we identified 9674 lincRNAs after removing those transcripts that are located within 1 kb from any known protein-coding genes (Supplemental file 3).
Previous studies in mammals have shown that lncRNAs are shorter, less conserved, and expressed at significantly lower level compared with protein-coding genes (Guttman et al. 2010; Cabili et al. 2011). To determine whether rainbow trout lincRNAs have similar features, we characterized the basic features of the identified lincRNAs by comparing them with protein-coding genes. We found that rainbow trout lincRNAs are on average about 1 s of the length of protein-coding genes (mean length of 705 nt for lincRNAs vs. 1635 nt for protein-coding transcripts) (Fig. 2a). Moreover, lincRNAs had fewer exons (on average, 1.3 exons for lincRNAs vs. 6.9 exons for protein-coding genes) (Fig. 2b). Notably, the mean length and average exon number of rainbow trout lincRNAs are shorter/smaller than those of human (∼1000 nt and 2.9 exons) (Cabili et al. 2011) and zebrafish (∼1000 nt and 2.8 exons). This could be due to the underestimation of the length and exon number of rainbow trout lincRNAs resulting from their lower abundance and lower sequencing depth (incomplete assembly). Furthermore, the expression levels of lincRNAs are on average about tenfold lower than those of protein-coding genes across 15 tissues (Fig. 3), which is consistent with the findings in human, mouse, and zebrafish (Cabili et al. 2011; Pauli et al. 2012; Guttman et al. 2010). Thus, the predicted rainbow trout lincRNAs share similar genomic features with lincRNAs from other species, suggesting that they are bona fide rainbow trout lincRNAs.

Structural characteristics of lincRNAs in comparison to protein-coding genes. a Cumulative distribution of transcript length for lincRNAs (red line) and protein-coding genes (blue line). Protein-coding genes larger than 8 kb were removed in the analysis. b Distribution of exon number for lincRNAs (red bars) and protein-coding genes (blue bars). Protein-coding genes with more than 20 exons were not included in the analysis

Comparison of expression levels of lincRNAs and protein-coding genes. Maximal expression abundance (log2-normalized FPKM counts estimated by Cufflinks) of each lincRNA (red solid line) and protein-coding gene (green broken line)
Analysis of Tissue-Specific Expression of Rainbow Trout lincRNAs
Recent studies have shown that lincRNAs are expressed in a more tissue-specific manner than protein-coding genes. We analyzed the expression pattern for each of the lincRNA transcripts. Of the 9674 potential lincRNAs, 8545 were expressed in more than one tissue (Fig. 4a, b; Supplemental file 4). The remaining 1129 lincRNAs displayed tissue-specific expression (Fig. 4d). Among the 15 tissues, the brain expressed the most number of tissue-specific lincRNAs (161), which is consistent with the result from a previous study in zebrafish (Kaushik et al. 2013). The skin, white muscle, and liver had relatively lower numbers of tissue-specific lincRNAs (Fig. 4c). The tissue specificity score for each lincRNA was calculated using an entropy-based metric that relies on Jensen-Shannon (JS) divergence (Cabili et al. 2011). Results showed that 46 % of rainbow trout lincRNAs were tissue-specific, relative to only 18 % of protein-coding genes (p < 10−16, Fisher exact test) (Fig. 5). Thus, rainbow trout lincRNAs exhibited more tissue specificity than protein-coding genes, which is in agreement with data from other species (Guttman et al. 2010; Cabili et al. 2011; Pauli et al. 2012).

Tissue-wise distribution of predicted lincRNAs. a Distribution of 9674 potential lincRNAs across 15 tissues. b Venn diagram representing 7783 lincRNAs in the gill (blue), intestine (yellow), kidney (orange), spleen (green), and stomach (pink). c Distribution of tissue-specific lincRNAs across 15 tissues. d Heatmap of 1129 tissue-specific lincRNAs across 15 tissues. Each column represents the expression levels of 1129 lincRNAs in the parent tissue vs. other tissues based on FPKM values

Tissue specificity of lincRNAs and protein-coding genes. Distribution of maximal tissue specificity scores calculated for each lincRNA (red solid line) or protein-coding transcript (green broken line) across all tissues
Tissue-specific expression of lincRNAs determined by computational analysis was validated by RT-PCR analysis. A total of 10 lincRNAs were selected for validation of their expression in 15 tissues. They include seven linRNAs specifically expressed in a particular tissue (Linc-OM9284 in the brain, Linc-OM8822 in the red muscle, Linc-OM8901 in the intestine, Linc-OM3900 in the stomach, Linc-OM8614 in the testis, Linc-OM8334 in fat, Linc-OM8318 in the kidney), two lincRNAs expressed in two tissues (Linc-OM8912 in oocyte and the skin, Linc-OM9283 in the skin and the liver), and one lincRNA ubiquitously expressed in all tissues (Linc-OM9274). As shown in Fig. 6, the RT-PCR result matches perfectly with the expression profiles estimated from deep sequencing data.

Validation of expression specificity of lincRNAs by RT-PCR analysis. Expression of ten selected lincRNAs was analyzed by RT-PCR in rainbow trout tissues including the brain (Br), oocyte (Oo), white muscle (Wm), pineal (Pi), fat (Fa), gill (Gi), skin (Sk), head kidney (Hk), testis (Te), spleen (Sp), stomach (St), liver (Li), red muscle (Rm), intestine (In), and kidney (Ki). 18S rRNA was used as a control for RNA quality
Co-expression of lincRNAs with Neighboring Coding Genes
The occurrence of pairs of neighboring lincRNA, protein-coding genes within expression clusters, suggests that such organization may be important for the regulatory function of lincRNAs (Cabili et al. 2011). Recent studies indicated that some lincRNAs may act in cis and regulate the expression of genes in their chromosomal neighborhood (Orom et al. 2010; Ponjavic et al. 2007; Luo et al. 2013; Cabili et al. 2011; Zhang et al. 2014). One expectation of the cis hypothesis is that the expression of lincRNAs and their neighboring genes would be correlated across all tissue samples. Therefore, we analyzed the expression patterns of 1146 (12 %) of identified lincRNAs that are located within 10 kb from a coding gene. We observed a more correlated expression pattern of lincRNAs and their neighboring coding genes (mean correlation: 0.211) compared to random coding gene pairs (mean correlation 0.042) [p < 2 × 10−16, Kolmogorov-Smirnov (KS) test] (Fig. 7). Meanwhile, lincRNAs: coding gene pairs also exhibited a modestly higher correlative expression pattern than coding gene pairs (mean correlation: 0.115) (p < 2.2 × 10−16, KS test). On the other hand, there is also a significant difference between neighboring coding gene pairs and random coding gene pairs (p < 7.9 × 10−13, KS test). This observation suggests that the correlation between lincRNAs and their neighbor coding genes are higher than both neighboring coding gene pairs and random coding gene pairs.

Correlation of expression patterns between pairs of neighboring genes. Shown are distributions of Pearson correlation coefficients in expression levels across the tissues between 1146 pairs of lincRNAs and their neighboring coding genes (green solid line), 9363 pairs of coding gene neighbors (blue broken line), and 8000 random pairs of protein-coding gene (red dotted line)
Functional Prediction of lincRNAs Based on Co-expression Network
The comprehensive lincRNA catalog allows us to investigate the potential functions of these novel transcripts in rainbow trout. Here, we built a co-expression network to associate lincRNAs with mRNAs by performing weighted gene co-expression network analysis (WGCNA) (Langfelder and Horvath 2008) and inferred the putative lincRNA functions based on “guilty-by-association” analysis. By clustering correlated genes together, we identified 34 co-expression gene modules containing 2963 lincRNAs and 10,321 protein-coding genes in total (Supplemental files 5 and 6). Notably, 6 of 34 modules are related to immune response, muscle differentiation, and neural development based on the enriched GO terms associated with their modules (Fig. 8).

Functional prediction of rainbow trout lincRNAs. a Upper panel, heatmaps showing expression patterns of all genes in each co-expression gene modules across 15 tissues. Middle panel, bar plots showing the corresponding module eigengene expression value. Lower panel, pie charts showing ratio of mRNAs and lincRNAs in each module. b Functional enrichment in each module. The length of bars indicates the significance (−log10 transferred FDR)
The functional annotations enriched in four modules (blue, grey60, tan, and green) are functionally related to immune responses (Fig. 8b and Supplemental file 7). In each of these four modules, we observed many lincRNAs that are highly expressed in the spleen, gill, and intestine (Fig. 8a), suggesting that these lincRNAs might be involved in immune-related processes. In the blue module, many genes were enriched in T cell receptor signaling and PI3K/AKT/mTOR signaling pathways (Supplemental file 8). The lincRNAs that are co-expressed with tyrosine-protein kinase (ITK), which phosphorylates PLCγ1 in T cell signaling (Andreotti et al. 2010), may play important roles in T cell signaling and function. PI3K and mTOR signaling pathways are important in regulating immune cell activation in neutrophils and mast cells and type I interferon production (Weichhart and Saemann 2008). Those lincRNAs that are co-expressed with PI3K or mTOR pathway genes are likely involved in these immune processes (Supplemental file 8). In grey60 module, the lincRNAs that are co-expressed with integrin, which mediates immune cells to penetrate into tissues (Evans et al. 2009), may play critical roles in immune cell migration and cell-cell interactions that occur during the course of an immune response. In the tan module, the lincRNAs that are co-expressed with Rab20, a key player in phagosome maturation (Pei et al. 2014), may function in phagocytosis. Likewise, lincRNAs in green module are co-expressed with MHC class I genes (Neefjes et al. 2011), indicating that they might be involved in processing and presenting antigen to T cells.
Genes in cyan module contains transcripts (165 protein-coding genes and 15 lincRNAs) that are highly expressed in muscle (Fig. 8). Most of the enriched genes in this module are related to functions or development of muscle (Supplemental file 7). Notably, the lincRNAs that are co-expressed with myoblast determination protein 2 (MyoD2) may play roles in regulating muscle differentiation. A previous study has demonstrated the role of a specific lncRNA in controlling muscle differentiation (Cesana et al. 2011).
Recent studies have shown that many lncRNAs are brain-specific, indicating their indispensable roles in brain development (Ng et al. 2012; Clark and Blackshaw 2014). This study also found that brain has the most tissue-specific lincRNAs (Fig. 4c). The lincRNAs in light yellow module are co-expressed with genes important for neural differentiation and development, such as dihydropyrimidinase-related protein (DRP) and Draxin precursor, indicating that they may function as important regulators of neurogenesis.
Collectively, the functional prediction analysis revealed that tissue-specific lincRNAs and protein-coding genes are enriched for processes specific to that tissue and essential in maintaining each tissue’s identity and functionality.
Conclusions
In this report, we provided the first comprehensive annotation of rainbow trout lincRNAs based on whole transcriptome sequencing of multiple tissues and identified 9674 novel lincRNA transcripts. These lincRNAs tend to be expressed in tissue-specific manner and share many characteristics with those in mammalian species. Co-expression network analysis suggested that many rainbow trout lincRNAs are associated with immune response, muscle differentiation, and neural development. The study lays the groundwork for future functional characterization of lincRNAs in rainbow trout.
References
Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick JS (2011) lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res 39:D146–D151.
Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ (2010) T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol 2:a002287.
Berthelot C, Brunet F, Chalopin D, Juanchich A, Bernard M, Noel B, Bento P, Da Silva C, Labadie K, Alberti A, Aury JM, Louis A, Dehais P, Bardou P, Montfort J, Klopp C, Cabau C, Gaspin C, Thorgaard GH, Boussaha M, Quillet E, Guyomard R, Galiana D, Bobe J, Volff JN, Genet C, Wincker P, Jaillon O, Roest Crollius H, Guiguen Y (2014) The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun 5:3657.
Bhartiya D, Kapoor S, Jalali S, Sati S, Kaushik K, Sachidanandan C, Sivasubbu S, Scaria V (2012) Conceptual approaches for lncRNA drug discovery and future strategies. Expert Opin Drug Discovery 7:503–513.
Cabianca DS, Casa V, Bodega B, Xynos A, Ginelli E, Tanaka Y, Gabellini D (2012) A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 149:819–831.
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25:1915–1927.
Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T, Bono H, Chalk AM, Chiu KP, Choudhary V, Christoffels A, Clutterbuck DR, Crowe ML, Dalla E, Dalrymple BP, De Bono B, Della Gatta G, Di Bernardo D, Down T, Engstrom P, Fagiolini M, Faulkner G, Fletcher CF, Fukushima T, Furuno M, Futaki S, Gariboldi M, Georgii-Hemming P, Gingeras TR, Gojobori T, Green RE, Gustincich S, Harbers M, Hayashi Y, Hensch TK, Hirokawa N, Hill D, Huminiecki L, Iacono M, Ikeo K, Iwama A, Ishikawa T, Jakt M, Kanapin A, Katoh M, Kawasawa Y, Kelso J, Kitamura H, Kitano H, Kollias G, Krishnan SP, Kruger A, Kummerfeld SK, Kurochkin IV, Lareau LF, Lazarevic D, Lipovich L, Liu J, Liuni S, Mcwilliam S, Madan Babu M, Madera M, Marchionni L, Matsuda H, Matsuzawa S, Miki H, Mignone F, Miyake S, Morris K, Mottagui-Tabar S, Mulder N, Nakano N, Nakauchi H, Ng P, Nilsson R, Nishiguchi S, Nishikawa S et al (2005) The transcriptional landscape of the mammalian genome. Science 309:1559–1563.
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I (2011) A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147:358–369.
Clark BS, Blackshaw S (2014) Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front Genet 5:164.
Conesa A, Gotz S (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008:619832.
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigo R (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22:1775–1789.
Evans R, Patzak I, Svensson L, De Filippo K, Jones K, Mcdowall A, Hogg N (2009) Integrins in immunity. J Cell Sci 122:215–225.
Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A (2008) The Pfam protein families database. Nucleic Acids Res 36:D281–D288.
Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:W29–W37.
Garber M, Grabherr MG, Guttman M, Trapnell C (2011) Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods 8:469–477
Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, Bateman A (2009) Rfam: updates to the RNA families database. Nucleic Acids Res 37:D136–D140.
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, Van De Vijver MJ, Sukumar S, Chang HY (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464:1071–1076.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458:223–227.
Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, Rinn JL, Lander ES, Regev A (2010) Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28:503–510.
Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, Attardi LD, Regev A, Lander ES, Jacks T, Rinn JL (2010) A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142:409–419.
Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, Wang Y, Kong B, Langerod A, Borresen-Dale AL, Kim SK, Van De Vijver M, Sukumar S, Whitfield ML, Kellis M, Xiong Y, Wong DJ, Chang HY (2011) Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet 43:621–629.
Kaushik K, Leonard VE, Kv S, Lalwani MK, Jalali S, Patowary A, Joshi A, Scaria V, Sivasubbu S (2013) Dynamic expression of long non-coding RNAs (lncRNAs) in adult zebrafish. PLoS One 8:e83616.
Khaitovich P, Kelso J, Franz H, Visagie J, Giger T, Joerchel S, Petzold E, Green RE, Lachmann M, Paabo S (2006) Functionality of intergenic transcription: an evolutionary comparison. PLoS Genet 2:e171.
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, Van Oudenaarden A, Regev A, Lander ES, Rinn JL (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A 106:11667–11672.
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G (2007) CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35:W345–W349.
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559.
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359.
Liao Q, Liu C, Yuan X, Kang S, Miao R, Xiao H, Zhao G, Luo H, Bu D, Zhao H, Skogerbo G, Wu Z, Zhao Y (2011) Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res 39:3864–3878.
Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH, Garber M, Curran M, Onder T, Agarwal S, Manos PD, Datta S, Lander ES, Schlaeger TM, Daley GQ, Rinn JL (2010) Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 42:1113–1117.
Luo H, Sun S, Li P, Bu D, Cao H, Zhao Y (2013) Comprehensive characterization of 10,571 mouse large intergenic noncoding RNAs from whole transcriptome sequencing. PLoS One 8:e70835.
Ma H, Hostuttler M, Wei H, Rexroad CE III, Yao J (2012) Characterization of the rainbow trout egg microRNA transcriptome. PLoS One 7:e39649.
Neefjes J, Jongsma ML, Paul P, Bakke O (2011) Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11:823–836.
Ng SY, Johnson R, Stanton LW (2012) Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J 31:522–533.
Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, Yagi K, Tomaru Y, Hasegawa Y, Nogami A, Schonbach C, Gojobori T, Baldarelli R, Hill DP, Bult C, Hume DA, Quackenbush J, Schriml LM, Kanapin A, Matsuda H, Batalov S, Beisel KW, Blake JA, Bradt D, Brusic V, Chothia C, Corbani LE, Cousins S, Dalla E, Dragani TA, Fletcher CF, Forrest A, Frazer KS, Gaasterland T, Gariboldi M, Gissi C, Godzik A, Gough J, Grimmond S, Gustincich S, Hirokawa N, Jackson IJ, Jarvis ED, Kanai A, Kawaji H, Kawasawa Y, Kedzierski RM, King BL, Konagaya A, Kurochkin IV, Lee Y, Lenhard B, Lyons PA, Maglott DR, Maltais L, Marchionni L, Mckenzie L, Miki H, Nagashima T, Numata K, Okido T, Pavan WJ, Pertea G, Pesole G, Petrovsky N, Pillai R, Pontius JU, Qi D, Ramachandran S, Ravasi T, Reed JC, Reed DJ, Reid J, Ring BZ, Ringwald M, Sandelin A, Schneider C, Semple CA, Setou M, Shimada K, Sultana R, Takenaka Y, Taylor MS, Teasdale RD, Tomita M, Verardo R, Wagner L, Wahlestedt C, Wang Y, Watanabe Y, Wells C, Wilming LG, Wynshaw-Boris A, Yanagisawa M et al (2002) Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420:563–573.
Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R (2010) Long noncoding RNAs with enhancer-like function in human cells. Cell 143:46–58.
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32:232–246.
Pang KC, Stephen S, Dinger ME, Engstrom PG, Lenhard B, Mattick JS (2007) RNAdb 2.0–an expanded database of mammalian non-coding RNAs. Nucleic Acids Res 35:D178–D182.
Pauli A, Valen E, Lin MF, Garber M, Vastenhouw NL, Levin JZ, Fan L, Sandelin A, Rinn JL, Regev A, Schier AF (2012) Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res 22:577–591.
Pei G, Repnik U, Griffiths G, Gutierrez MG (2014) Identification of an immune-regulated phagosomal Rab cascade in macrophages. J Cell Sci 127:2071–2082.
Perkel JM (2013) Visiting “noncodarnia”. Biotechniques 54(301):303–304.
Ponjavic J, Ponting CP, Lunter G (2007) Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res 17:556–565.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129:1311–1323.
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M (2011) pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12:77.
Salem M, Rexroad CE, Wang J, Thorgaard GH, Yao J (2010a) Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics 11:564
Salem M, Xiao C, Womack J, Rexroad CE 3rd, Yao J (2010b) A microRNA repertoire for functional genome research in rainbow trout (Oncorhynchus mykiss). Mar Biotechnol (NY) 12:410–429.
Salem M, Paneru B, Al-Tobasei R, Abdouni F, Thorgaard GH, Rexroad CE, Yao J (2015) Transcriptome assembly, gene annotation and tissue gene expression atlas of the rainbow trout. PLoS One 10:e0121778.
Solda G, Makunin IV, Sezerman OU, Corradin A, Corti G, Guffanti A (2009) An Ariadne’s thread to the identification and annotation of noncoding RNAs in eukaryotes. Brief Bioinform 10:475–489.
Sun L, Zhang Z, Bailey TL, Perkins AC, Tallack MR, Xu Z, Liu H (2012) Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC Bioinformatics 13:331.
Sun K, Zhao Y, Wang H, Sun H (2014) Sebnif: an integrated bioinformatics pipeline for the identification of novel large intergenic noncoding RNAs (lincRNAs)—application in human skeletal muscle cells. PLoS One 9:e84500.
Thorgaard GH, Bailey GS, Williams D, Buhler DR, Kaattari SL, Ristow SS, Hansen JD, Winton JR, Bartholomew JL, Nagler JJ, Walsh PJ, Vijayan MM, Devlin RH, Hardy RW, Overturf KE, Young WP, Robison BD, Rexroad C, Palti Y (2002) Status and opportunities for genomics research with rainbow trout. Comp Biochem Physiol B Biochem Mol Biol 133:609–646
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515.
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, Wysocka J, Lei M, Dekker J, Helms JA, Chang HY (2011) A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472:120–124.
Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W (2013a) CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res 41:e74.
Wang L, Tripurani SK, Wanna W, Rexroad CE 3rd, Yao J (2013b) Cloning and characterization of a novel oocyte-specific gene encoding an F-Box protein in rainbow trout (Oncorhynchus mykiss). Reprod Biol Endocrinol 11:86.
Weichhart T, Saemann MD (2008) The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann Rheum Dis 67(Suppl 3):iii70–4.
Zhang K, Huang K, Luo Y, Li S (2014) Identification and functional analysis of long non-coding RNAs in mouse cleavage stage embryonic development based on single cell transcriptome data. BMC Genomics 15:845.
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322:750–756.
Zhu Y, Yu M, Li Z, Kong C, Bi J, Li J, Gao Z (2011) ncRAN, a newly identified long noncoding RNA, enhances human bladder tumor growth, invasion, and survival. Urology 77:510.e1–5.
Acknowledgments
This study was supported by the USDA ARS Cooperative Agreement No. 58-1930-0-059. It is published with the approval of the director of the West Virginia Agricultural and Forestry Experiment Station as scientific paper No. 3268.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplemental file 1
Primers used for validation of expression of 10 selected lincRNAs. (XLSX 9 kb)
Supplemental file 2
Total transcript counts in each step of lincRNA prediction pipeline. (XLSX 10 kb)
Supplemental file 3
Catalog of 9764 transcribed lincRNAs in rainbow trout. (XLSX 2309 kb)
Supplemental file 4
Venn diagram representing rainbow trout lincRNAs across different tissues. (A) Overlapping expression profiles of lincRNAs across testis (blue-Te), oocyte (yellow-Oo), pineal (orange-Pi), brain (green-Br), and liver (pink-Li). (B) Overlapping expression profiles of lincRNAs across white muscle (blue-Wm), red muscle (yellow-Rm), skin (orange-Sk), fat (green-Fa) and head kidney (pink-Hk). (JPG 142 kb)
Supplemental file 5
Hierarchical clustering dendrogram of lincRNAs together with protein coding genes. (JPG 50 kb)
Supplemental file 6
Genes and their memberships to each module. MM stands for module membership, which is the correlation between a gene and a module. PMM is the p-value of MM. (XLSX 170 kb)
Supplemental file 7
GO analysis of modules. GO terms enriched in specific modules were tested by Fisher’s exact test. (XLSX 170 kb)
Supplemental file 8
KEGG pathway analysis and network of hub gene of blue module. (A) and (B) Hub gene network. (C) mTOR signaling pathway. (D) T cell receptor signaling pathway. (E) PI3K-AKT signaling pathway. (JPG 709 kb)
Rights and permissions
About this article

Cite this article
Wang, J., Fu, L., Koganti, P.P. et al. Identification and Functional Prediction of Large Intergenic Noncoding RNAs (lincRNAs) in Rainbow Trout (Oncorhynchus mykiss). Mar Biotechnol 18, 271–282 (2016). https://doi.org/10.1007/s10126-016-9689-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-016-9689-5