
Abstract
A chymotrypsin was purified from the gastric juice of California spiny lobster (Panulirus interrutpus), using preparative electrophoresis and affinity chromatography on agarose-p-aminobenzamidine. The molecular mass was estimated by polyacrylamide gel electrophoresis (SDS-PAGE) under denaturing conditions to be 28 kDa. Chymotrypsin activity was totally inhibited by phenylmethylsulfonyl fluoride (PMSF) and chymostatin. Lobster chymotrypsin had optimal pH 7.0–8.0 and temperature of 55 °C. The enzyme is highly stable under a wide range of pH (retaining up to 80 % of activity after 1 h of incubation at pH 3.0, 5.0, and 12.0), showing higher stability at pH 8.0, and was inactivated after 20 min at 55 °C. Lobster chymotrypsin was able to hydrolyze protein substrates at as low as pH 3.0. These results are consistent with the findings of enzyme stability. Activity was assessed after incubation of enzyme with different organic solvents (in the range of 10–50 %); when tested in the presence of acetone, ethanol, propanol, and butanol, lobster chymotrypsin residual activity was >80 %; whereas in the presence of dimethyl sulfoxide (DMSO) and toluene, lobster chymotrypsin residual activity was <80 %. Deduced amino acid sequence, corroborated by mass spectrometry, was determined.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proteolytic enzymes from the digestive system of crustacean decapods have been the subject of extensive work. Serine, cysteine, aspartic, and metallo digestive peptidases have been found in species in this order. Serine proteinases, trypsin, and chymotrypsin are the most common and abundant digestive proteinases in crustacean decapods studied so far (Muhlia-Almazán and García-Carreño 2002), including California spiny lobster Panulirus interruptus (Celis-Guerrero et al. 2004) and Caribbean spiny lobster Panulirus argus (Perera et al. 2008a, b), Pacific whiteleg shrimp Litopenaeus vannamei (Hernández-Cortés et al. 1997) and giant tiger prawn Penaeus monodon (Tsai et al. 1991), and warrior swimming crab Callinectes bellicosus and arched swimming crab Callinectes arcuatus (Díaz-Tenorio et al. 2006). Crustacean serine peptidases appear to respond to changes in the molt cycle and starvation (Muhlia-Almazán and García-Carreño 2002; Perera et al. 2008b), diet protein sources (Perera et al. 2005), and innate immune system (Shi et al. 2008). This information is important to understand the physiology of protein digestion and particular nutritional needs, which is of major concern in ecology and potential farming. Investigation of structural, biochemical, and operational parameters of these enzymes could lead to maximizing use of these marine resources (Castillo-Yáñez et al. 2006; Klomklao 2008).
Chymotrypsin (EC 3.4.21.2), a serine peptidase, has been widely studied to determine protein structure, activation, and biochemical properties; however, most research has been restricted to mammals, specifically to cattle (Bos taurus). In cattle, chymotrypsin is the second major peptidase of pancreatic juice, where it is found as chymotrypsinogen. Activation of chymotrypsin is regulated by limited proteolysis by trypsin in the duodenum lumen (Gráf et al. 2013). Amino acid residues Ser195, His57, and Asp102 form the catalytic triad that are located at the entrance of the substrate-binding pocket and are highly conserved in the sequences of peptidases of the S1 family (Perona and Craik 1997; Gráf et al. 2013). Chymotrypsins hydrolyze peptide bonds on the carboxylic side of Phe, Tyr, Trp, and Leu (Di Cera 2009). During hydrolysis, the Ser195 amino acid residue attacks the carbonyl of the scissile peptide bond, assisted by His57-H+, which is stabilized by a hydrogen bond with Asp102 (Hedstrom 2002). In general, the activity of chymotrypsins is limited to neutral-to-alkaline pH because the pK a of the His57 amino acid residue is ~7.0 (Hedstrom 2002) and deprotonation is needed to initiate the nucleophilic attack on the peptide bond.
Chymotrypsin from a number of marine species has been purified and characterized (Raae and Walther 1989; Kristjansson 1991; Tsai et al. 1991; Eberhardt 1992; Van Wormhoudt et al. 1992; Heu et al. 1995; Le Chevalier et al. 1995; Roy et al. 1996; Hernández-Cortés et al. 1997; Fong et al. 1998; Castillo-Yañez et al. 2004, 2009; Yang et al. 2009; El Hadj et al. 2010; Balti et al. 2012; Navarrete-del-Toro et al. 2015). Fish chymotrypsins are the most studied. Less attention has been given to chymotrypsin of marine invertebrates. The optimum pH for proteolytic activity in most digestive extracts from crustacean decapods is alkaline; however, the gastric juice is slightly acidic, ranging from pH 4.0 to 6.0 (Navarrete Del Toro et al. 2006). This is consistent with aspartic and cysteine digestive peptidases having an acid optimal pH values in some decapods (Teschke and Saborowski 2005; Rojo et al. 2010a) and with serine peptidases bearing unique characteristics, such as activity and/or stability at acidic pH (Hernández-Cortés et al. 1997; Celis-Guerrero et al. 2004).
The peptidases in the digestive tract of P. interruptus have been characterized in terms of activity and stability of catalytic types, pH, and temperature (Celis-Guerrero et al. 2004). The composition of digestive peptidases is similar in the gastric juice and the midgut gland extract. Peptidase activity was detected in the pH range of 2.0–12.0 and showed an optimal pH of 8.0–9.0 (Celis-Guerrero et al. 2004). At pH 8.0, the main peptidase activity is related to trypsin and chymotrypsin, as detected by the synthetic substrates succinyl-L-Ala-Ala-Pro-L-Phe-p-nitroanilide (SAPNA) and benzoyl-Arg-p-nitroanilide (BAPNA), by proteolytic activity on polyacrylamide gels (S-SDS-PAGE) and the effect of inhibitors at pH 8.0. Activity in gastric juice at acid pH results from aspartic and serine peptidases (Celis-Guerrero et al. 2004; Navarrete Del Toro et al. 2006); however, these peptidases have not been characterized so far.
Because the pH of the gastric juice of P. interruptus is slightly acidic (Navarrete Del Toro et al. 2006) and serine peptidases are by far the most abundant peptidases, we hypothesize that digestive serine peptidases from lobster bear characteristics of activity and stability at slightly acidic pH values. To probe this, we purified a digestive peptidase from the gastric juice of lobster and determined its identity by using peptidase inhibitors, mass spectrometry, and gene sequencing. We also investigated its functional parameters, including effect of pH and temperature on enzyme activity and stability, effect of pH on the activity towards proteinaceous substrates, effect of solvents on enzyme activity and kinetic parameters, and the capacity to hydrolyze varied proteins.
Materials and Methods
Preparation of Enzyme Extract
Live adult California spiny lobsters, P. interruptus, were obtained from the local market and maintained in aquaria at 20 °C in a constant marine water flow and fed with squid muscle. The gastric juice was extracted from live lobsters (Navarrete del Toro et al. 2006). In brief, a catheter consisting of a syringe with a flexible tube adapted to the tip was inserted into the lobster’s oral cavity, and the gastric juice was removed by aspiration. The gastric juice was centrifuged at 10,000×g for 10 min at 4 °C. The supernatant was collected, lyophilized, and stored at −20 °C until used. The supernatant was analyzed for protein concentration, following the method of Bradford (1976), using bovine serum albumin (BSA; B4287, Sigma-Aldrich, St. Louis, MO) as the standard.
Determination of Chymotrypsin Activity
Chymotrypsin activity was determined using the chromogenic substrate SAPNA (S7388, Sigma-Aldrich); 10 μL enzyme solution were placed in a microplate well, and the reaction was initiated by adding 290-μL 0.1-mM SAPNA in 50 mM Tris–HCl at pH 8.0, containing 20 mM CaCl2. Production of p-nitroaniline was determined by measuring increased absorbance at 410 nm in a microplate reader (VersaMax, Molecular Devises, Sunnyvale, CA) every 30 s for 5 min at 25 °C. Specific activity was calculated as: units per milligram = (Abs 410 nm min−1 × reaction volume in mL)/(8800 × mg of enzyme). Trypsin activity was monitored during the purification process by using BAPNA (B4875, Sigma-Aldrich) as the substrate, following the method described for determining chymotrypsin activity. All determinations were done in triplicate.
Purification of Chymotrypsin
To purify chymotrypsin, 8 mg protein from the gastric juice were mixed with 2× sample buffer (0.125 M Tris–HCl at pH 6.8, 20 % v/v glycerol, 0.02 % w/v bromophenol blue) and loaded into a preparative electrophoresis system (PrepCell, BioRad Laboratories, Hercules, CA) connected to a fraction collector system (GradiFrac, GE Healthcare, Little Chalfont, UK). A 12 % polyacrylamide gel (5 cm diameter and 8 cm high) was used. The electrode buffer consisted of 5 mM Tris and 38 mM glycine at pH 8.3. Electrophoresis was run at a constant 12 W. One-milliliter fractions were collected, and all fractions were determined for trypsin and chymotrypsin activity. Fractions containing chymotrypsin activity were pooled and concentrated by centrifugation using a centrifuge filter unit of 10 kDa cutoff at 5000×g at 4 °C (EMD Millipore, Billerica, MA). After concentration, affinity chromatography on a 1-mL agarose-p-aminobenzamidine matrix (A-8332, Sigma-Aldrich) was run. Then, 40 μg of protein was suspended in an equilibrium buffer (0.1 M Tris–HCl, pH 7.0) and loaded into the previously equilibrated column. The proteins bound to the column were gradually eluted with 10-mL 0.1 M Tris–HCl at pH 7.0; 10-mL 0.1 M Tris-HCl at pH 7.0, containing 0.5 M NaCl; and then with 5-mL 0.1 M Tris–HCl at pH 7.0, containing 0.5 M butylamide. Lastly, the column was washed with 5-mL 0.1-M sodium acetate at pH 4.5, containing 0.5 M NaCl and re-equilibrated.
Protein Electrophoresis and Substrate-SDS-PAGE
SDS polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to Laemmli (1970). Samples were mixed with 2× sample buffer (0.125 M Tris–HCl at pH 6.8, 4 % (w/v) SDS, 20 % (v/v) glycerol, 0.02 % (w/v) bromophenol blue). Samples were incubated at room temperature or at 100 °C for 10 min prior to electrophoresis. For reducing conditions, samples were mixed with 2× sample buffer containing 0.2 M dithiothreitol (DTT) and incubated at 100 °C for 10 min. Samples were loaded into a 12 % polyacrylamide gel. Electrophoresis was run at 15 mA/gel at 4 °C, using a SE-260 mini-vertical electrophoresis unit (GE Healthcare). After electrophoresis, the gels were stained for 2 h with staining solution (0.05 % (w/v) Commmassie Brilliant Blue R-250, 7 % (v/v) acetic acid, 40 % (v/v) methanol). Proteins were revealed by soaking for 2 h in destaining solution (7 % (v/v) acetic acid and 40 % (v/v) methanol).
Substrate-SDS-PAGE (S-SDS-PAGE) was conducted to visualize active peptidases in 12 % polyacrylamide gels (García-Carreño et al. 1993). Gels were immersed in substrate solution containing 0.25 % hemoglobin (H-2625, Sigma-Aldrich) in 50 mM glycine-HCl at pH 3.0 or 3 % casein (C-7078, Sigma-Aldrich) in 50 mM Tris–HCl at pH 8.0, and incubated at 4 °C for 30 min. The gels were soaked in substrate solution at room temperature for 90 min. After the incubation time, the gels were washed with distilled water and stained for 2 h with staining solution. The gel was immersed in destaining solution for 2 h.
Inhibition Assays
Pure enzyme was incubated for 1 h at room temperature with the following inhibitors: 1 mM PMSF (P-7626, Sigma-Aldrich), 1 mM pepstatin A (P-4265, Sigma-Aldrich), 1 mM E-64 (E-3132, Sigma-Aldrich), 1 mM tosyl-l-lysine chloromethyl ketone (TLCK; T-7254, Sigma-Aldrich), 1 mM tosyl-l-phenylalanine chloromethyl ketone (TPCK; T-4376, Sigma-Aldrich), 20 µM chymostatin (C-7268, Sigma-Aldrich), and 1:10 dimethyl sulfoxide (DMSO) as the control. After incubation, chymotrypsin activity and S-SDS-PAGE were determined as described earlier. Total protein used per assay was 0.375 μg.
Optimal Temperature and pH
To determine optimal pH, chymotrypsin activity was assessed at pH: 5.0, 6.0, 7.0, 8.0, 9.0, and 10.0, as described above, with the exception that reaction buffer was replaced by a universal buffer containing 57 mM boric acid, 36 mM citric acid, and 38 mM monosodic∙H2O phosphate; the pH was adjusted with HCl (Stauffer 1989). To determine optimal temperature, chymotrypsin activity was assessed at 15, 25, 35, 45, 55, and 65 °C at pH 8.0.
pH and Thermal Stability
To determine the effect of pH on stability, pure enzyme (2 μg) was mixed in 320 μL of the universal buffer at pH 3.0, 5.0, 8.0, and 12.0 and incubated at 25 °C; 50-μL aliquots of this mixture were withdrawn at 0, 20, 40, and 60 min of incubation, pH was adjusted to 8.0 with 200 mM Tris–HCl (pH 8.0), and the resulting mixture was used to measure chymotrypsin activity, as described earlier.
To determine temperature effect on stability, pure enzyme (2 μg) was mixed in 320-μL 50-mM Tris–HCl at pH 8.0, containing 20 mM CaCl2, and incubated at 25, 35, 45, and 55 °C; 50-μL aliquots of this mixture were withdrawn at 0, 20, 40, and 60 min incubation. Chymotrypsin activity was measured, as described earlier.
Hydrolysis of Proteins
To determine the pH range for proteinolytic activity of the purified enzyme, BSA (B-4287, Sigma-Aldrich) and collagen type VII from rat tail (C-8897, Sigma-Aldrich) were hydrolyzed within the range of pH 3.0 to 8.0. Pure enzyme (0.12 μg) was mixed in 10-μL 4× universal buffer adjusted to the various pH values. Proteinaceous substrates (BSA or rat collagen, final concentration of 6 mg mL-1) were mixed with enzyme. The mixtures were incubated for 24 h at 25 °C. The reactions were stopped by adding loading buffer containing DTT and incubated for 10 min at 100 °C. Hydrolysis products were analyzed by 12 % SDS-PAGE.
Effect of Solvents on Enzyme Activity
To determine the effect of solvents on enzyme activity, enzyme (0.85 μg) was incubated for 30 min at room temperature with ethanol (1.00983, Merck Millipore, Billerica, MA), propanol (I-9516, Sigma, Aldrich), n-butanol (537993, Sigma-Aldrich), DMSO (D-5879, Sigma-Aldrich), acetone (A-4206, Sigma-Aldrich), or toluene (155004, Sigma-Aldrich). Solvents were assayed at 10, 30, and 50 % (v/v) concentration. After incubation, enzyme activity was measured, as described earlier. Residual activity was compared with that of the enzyme without solvents. To discard the effect of solvents over substrate stability, a control assay was run without the enzyme.
Kinetic Studies
Kinetic parameters of the purified chymotrypsin were assayed by steady-state enzyme kinetics, using SAPNA as the substrate. Reaction rate was calculated in micromoles of p-nitroaniline liberated per minute of reaction. Substrate concentration ranged from 0.03 to 0.3 μM. Enzyme concentration in the reaction was 1.96 μg mL−1. The Michaelis–Menten constant and the maximal velocity were calculated by non-linear fit analysis, using Prism 5 software (Graphpad Software, San Diego, CA). The catalytic constant (k cat) was calculated by dividing the maximal velocity by the enzyme molar concentration in the reaction, calculated by molecular weight.
Statistical Analyses
The results of determining enzyme activity under different treatments were analyzed by parametric statistics. One-way ANOVA was performed, followed by Tukey’s test. Statistical significance was set at P < 0.05.
Mass Spectrometry Analysis
For identification, 2 μg protein was separated by 12 % SDS-PAGE. After electrophoresis, the gel was stained, as described earlier. The protein band was cut and extracted from the gel. The gel band was destained, reduced with 10 mM DTT, and alkylated with 55 mM iodoacetamide. It was then tryptic digested before analysis by nano-LC-MS/MS at the TSRI Center for Mass Spectrometry, La Jolla, CA.
Chymotrypsin cDNA Sequencing
Total RNA was isolated from the midgut gland of the lobster using TRIzol® reagent (10296–010, Ambion, Life Technologies, Carlsbad, CA). After treating the sample with DNAse, complementary DNA (cDNA) was synthesized from 1 μg total RNA using the reverse transcription system kit (A-3500, Promega, Madison, WI) and the Oligo(dT)15 primer.
A set of primers that hybridize in highly conserved regions of known decapod chymotrypsin messenger RNA (mRNA) sequences were designed. A 681-pb region of California spiny lobster chymotrypsin cDNA was amplified by PCR. The reaction included 6 μL polymerase (GoTaq Green Master Mix (M7121, Promega), 1 μL (10 μM) of each primer ChyF (3′-GCCAGCGGAAACCCGC-5′) and ChyR2 (3′-GGCATCACCTCCTTCGG-5′), 100 ng of lobster cDNA, and water to a final volume of 12 μL. The reaction temperatures were 95 °C for 3 min for initial denaturation, 30 cycles at 95 °C for 30 s, 56 °C for 30 s, 68 °C for 1 min, and a final extension at 72 °C for 10 min. The PCR reactions were visualized in 1 % agarose gels. The single band that was produced was sequenced in both directions, using the same set of primers.
To obtain the full-length sequence of lobster chymotrypsin transcript, we used a cDNA amplification kit (SMART race, 634914, Clontech Laboratories, Mountain View, CA). First-strand cDNA synthesis of 3′RACE-ready and 5′RACE-ready were performed separately from 1 μg poly A+ lobster RNA obtained with a mRNA kit (Oligotex, 70022, Qiagen, Hilden, Germany). For PCR amplification of lobster 3′and 5′ chymotrypsin cDNA ends, the Universal primers included in the SMART race kit and the ChyF and ChyR2 primers were used, with the template 3′RACE-ready and 5′RACE-ready cDNA, respectively. Both reactions produced a single PCR band that was used for ligation in a sequencing vector (pGEM-Teasy, A1360, Promega) that was cloned into Escherichia coli DH5α-competent cells (18265–017, Invitrogen, Carlsbad, CA) following standard cloning methods. Plasmid DNA was isolated from three positive colonies using the QIAprep Spin Miniprep Kit (27104, Qiagen) and used for sequencing reactions.
Results
Purification of Chymotrypsin
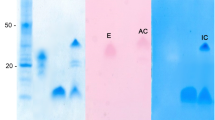
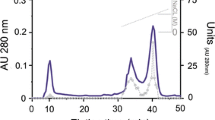
A chymotrypsin was purified from the gastric juice of the lobster by preparative electrophoresis and affinity chromatography on an agarose-p-aminobenzamidine matrix. In the preparative electrophoresis, chymotrypsin activity appeared as a single peak; nonetheless, all these fractions also included trypsin activity (Fig. 1a). The fractions containing chymotrypsin activity were pooled and concentrated, and the SDS-PAGE (Fig. 1a, lanes 1 and 2) and S-SDS-PAGE (Fig. 1a, lanes 3 and 4) analyses showed that concentrated fractions contained several protein and activity bands. Affinity chromatography separated chymotrypsin from trypsin activity (Fig. 1b). Chymotrypsin activity appeared as a single band by S-SDS-PAGE (Fig. 1b, lane 5), while the trypsin-containing fraction had several activity bands by S-SDS-PAGE (Fig. 1b, lane 6). Fraction 12 from affinity chromatography was concentrated by ultrafiltration and used in the characterization analysis. Table 1 is a summary of the purification process; the overall purification factor was 13.11, and the yield was 10 %.

Proteins in the gastric juice of California spiny lobster (Panulirus interruptus) were separated by preparative electrophoresis. a Trypsin and chymotrypsin activity in eluted fractions was monitored. Fractions containing chymotrypsin from preparative electrophoresis were concentrated and analyzed by SDS-PAGE and S-SDS-PAGE at pH 8.0 (lanes 1 and 3, gastric juice; lanes 2 and 4, pooled fractions) and submitted to affinity chromatography on a p-aminobenzamidine matrix. b The sample was equilibrated using 0.1 M Tris–HCl at pH 7.0, then chymotrypsin activity was eluted using 0.1 M Tris–HCl and 0.5 M NaCl at pH 7.0, and trypsin activity was eluted using 0.1 M Tris–HCl and 0.5 M butylamide at pH 7.0. Fractions containing chymotrypsin (lane 5) and trypsin (lane 6) were analyzed by S-SDS-PAGE at pH 8.0
Characterization of Chymotrypsin
Relative Molecular Weight
Lobster chymotrypsin was analyzed by SDS-PAGE, appearing as a single band under all electrophoretic conditions, but migrating at different rates. Lobster chymotrypsin appeared at 20.1 kDa when it was not heat-treated prior to electrophoresis (Fig. 2, lane 1). When lobster chymotrypsin was incubated at 100 °C for 10 min with and without DDT, the protein appeared at 28 kDa (Fig. 2, lanes 2 and 3). Since the enzyme appeared as a single band in the DTT/heat-treated sample, it indicates that lobster chymotrypsin is composed of a single polypeptide chain.

Electrophoretic analysis of lobster chymotrypsin. Lobster chymotrypsin was incubated at room temperature for 10 min and analyzed by SDS-PAGE (lane 1), incubated at 100 °C for 10 min and analyzed by SDS-PAGE (lane 2), and treated with DTT, at 100 °C for 10 min and analyzed by SDS-PAGE (lane 3)
Effect of Inhibitors
The purified enzyme exclusively hydrolyses the synthetic substrate SAPNA, but not BAPNA. The residual activity was determined after incubation with a series of inhibitors. The results are presented in Fig. 3. The enzyme lost 95 % of activity by the effect of PMSF. In contrast, E-64 and pepstatin A had no effect on enzyme activity. Trypsins and chymotrypsins are the major peptidases in the gastric juice of the lobster (Celis-Guerrero et al. 2004); therefore, we used TLCK, TPCK, and chymostatin to distinguish whether the purified enzyme is a chymotrypsin or trypsin. TLCK (10 %) and TPCK (25 %) barely inhibited enzyme activity, while chymostatin completely abolished it (Fig. 3a).

Activity of lobster chymotrypsin was determined after incubation with different inhibitors for 1 h. a Mean values of residual activity carried out in triplicate, using SAPNA as substrate. Datasets that were significantly different (P < 0.05) from each other are labeled with different letter codes. b Activity was determined by S-SDS-PAGE at pH 3.0, using hemoglobin as substrate and c by S-SDS-PAGE at pH 8.0, using casein as substrate
We determined the enzyme proteinolytic activity at pH 3.0 and pH 8.0 by S-SDS-PAGE. An activity band of ~20.1 kDa was found under both conditions; hence, we performed an inhibition assay to determine whether lobster chymotrypsin is the enzyme responsible for the activity. The chymotrypsin was incubated with PMSF, pepstatin A, E-64, TLCK, TPCK, and chymostatin, and then was assayed for proteinolytic activity by S-SDS-PAGE at pH 3.0 and 8.0. While PMSF completely abolished the activity of lobster chymotrypsin, pepstatin A, E-64, TLCK, and TPCK did not affect activity, and chymostatin slightly affected enzyme activity (Fig. 3b, c). These results are similar to those obtained by hydrolysis of SAPNA and using the same set of inhibitors (Fig. 3a). This indicates that lobster chymotrypsin is responsible for the activity under acidic and alkaline conditions.
Effect of pH on Enzyme Activity and Stability
The effect of pH on enzyme activity and stability using SAPNA as the substrate was determined. The optimal pH for hydrolysis of SAPNA is between 7.0 and 8.0; activity decreases at pH 5.0 (Fig. 4a). Lobster chymotrypsin conserves 100 % of its activity after 1 h at pH 8.0 and conserves more than 80 % of its activity after 1 h at pH 3.0, 5.0, and 12.0 (Fig. 4b).

Effect of pH on lobster chymotrypsin a activity and b stability, and effect of temperature on lobster chymotrypsin c activity and d stability. Activity was determined with SAPNA as substrate and results are given as the mean of triplicate assays. Datasets that were significantly different (P < 0.05) from each other at a given time are indicated with an asterisk
Effect of pH on Protein Hydrolysis
We investigated the capability of lobster chymotrypsin to hydrolyze BSA and collagen in the pH range from 3.0 to 8.0. Lobster chymotrypsin hydrolyzes both substrates in the entire range, exhibiting optimal hydrolysis at alkaline pH (Fig. 5a, b). These results are similar to those obtained by S-SDS-PAGE and demonstrate that lobster chymotrypsin is able to hydrolyze proteins under a wide range of pH.

Protein hydrolysis by lobster chymotrypsin. Lobster chymotrypsin was incubated in the pH range from 3.0 to 8.0 at room temperature for 20 h, with a bovine serum albumin and b rat tail collagen. After incubation, the reaction was stopped with the sample buffer and boiled for 10 min. The products of the reactions were analyzed by SDS-PAGE
Effect of Temperature on Enzyme Activity and Stability
The effect of temperature on enzyme activity and stability is depicted in Fig. 4c, d. The apparent optimal temperature for hydrolysis of SAPNA is 55 °C; activity drops to 25 % at 65 °C. This indicates that lobster chymotrypsin is rapidly inactivated at temperatures above 55 °C, which is consistent with our findings of enzyme stability. Lobster chymotrypsin conserves 83, 67, and 54 % of activity after 1 h of incubation at 25, 35, and 45 °C, respectively. The enzyme loses 88 % of its activity within the first 20 min at 55 °C and is completely denatured within 40 min at 55 °C. Hence, the rapid decline in activity at 65 °C is an indicator of low stability at temperatures above 55 °C.
Effect of Solvents on Enzyme Activity
Solvents affect the stability of lobster chymotrypsin (Fig. 6). Acetone significantly reduces residual activity to ~80 % at all tested concentrations. Propanol reduced the residual activity to ~80 % when tested at concentrations of 10 and 30 %. Activity was not affected using 50 % propanol. Ethanol and butanol did not significantly diminished chymotrypsin activity at any tested concentration. DMSO and toluene affected activity in a greater extent. When chymotrypsin was incubated with 10 % DMSO, activity decreased to 85 %, while 30 % DMSO reduced the activity to ~70, and 50 % DMSO reduced the activity to ~45 %. Toluene reduced activity to ~70 % when tested at concentrations of 10 and 30 %, while 50 % toluene reduced the activity to ~60 %.

Effect of solvents on enzyme activity. Lobster chymotrypsin was incubated with solvents for 30 min at room temperature, and activity was assayed with SAPNA as substrate. Results are given as the mean of triplicate assays. Datasets that were significantly different (P < 0.05) from each other are indicated with different letter codes
Kinetic Parameters
Kinetic constants of lobster chymotrypsin were assessed by hydrolysis of the substrate SAPNA. The values for K m , k cat, and k cat /K m are 0.28 mM, 1.07 s−1 and 3.8 s−1 mM−1, respectively (Table 2).
Deduced Amino Acid Sequence Analysis
The full-length nucleotide 816-pb sequence encodes for 271-amino acids of the P. interruptus chymotrypsinogen (GenBank ID, KP675948) from the ATG start codon through the TGA stop codon; sequence analysis predicts a putative activation peptide of 45-amino acids leaving an active protein of 226-amino acids. This configuration was inferred by protein alignment with crustacean chymotrypsinogens, for which the N-terminus sequence has been determined (Fig. 6, IVGGVA of yellowleg shrimp, Navarrete-del-Toro et al. 2015).
At least three peptide sequences obtained by mass spectrometry matched with the deduced amino acid sequence (Fig. 7, dotted box). Additionally, the theoretical molecular weight for the deduced mature protein is 24.0 kDa, a value similar to that obtained experimentally (Fig. 2, lanes 2 and 3). This confirmed the identity of P. interruptus chymotrypsin. The calculated isoelectric point is 5.54. BLASTp analysis of the deduced amino acid sequence P. interruptus chymotrypsin revealed high homology to chymotrypsins of closely related decapod species (69–79 % of identity) and 36 % to cattle chymotrypsinogen (Fig. 7). The P. interruptus chymotrypsin deduced amino acid sequence contains the serine protease catalytic triad residues Ser195, His57, and Asp102 (cattle chymotrypsinogen numbering, bold and shaded in Fig. 7) and showed the same residues as the brachyurin from Uca pugilator, forming the S1 binding pocket (G189, G216, and D226) (Perona and Craik 1995).

Alignment of amino acid sequence of serine peptidases. Chymotrypsin from Panulirus interruptus (GeneBenk ID, KP675948); chymotrypsin from Litopenaeus vannamei (GenBank ID, CAA71673), Farfantepenaeus californiensis (GenBank ID: AGR49093), and Scylla paramamosain (GenBank ID: AEE25770); collagenase from Uca pugilator (GenBank ID: AAC47030); and chymotrypsinogen from Bos taurus (GenBank ID: XP_003583409). The percentage of identity of P. interruptus sequence to each serine peptidase is indicated in percentage. Chymotrypsinogen numbering is used. Matched peptides found by MS are in dotted boxes. Residues forming the catalytic triad are bold shaded. Important residues of the S1 binding pocket are bold. Lines between cysteine residues indicate conserved disulfide bridges, and asterisks indicate non-conserved cysteine residues
Discussion
The serine peptidases, trypsin and chymotrypsin, are the major peptidases in the gastric juice of California spiny lobster P. interruptus (Celis-Guerrero et al. 2004), and the milieu of the gastric juice is slightly acidic (Navarrete Del Toro et al. 2006). We purified and characterized a chymotrypsin from the gastric tract of the lobster to determine whether it exhibits activity or stability at an acidic pH. For purification, we submitted the gastric juice to preparative electrophoresis, which allowed us to separate proteins by means of molecular weight and selected the fractions containing the highest amount of chymotrypsin activity. However, these fractions also contained trypsin activity. The chymotrypsin was separated from trypsins by means of affinity chromatography in p-aminobenzamidine; the differential elution of lobster chymotrypsin and trypsins is an indicator of a different mode and strength of binding. The same pattern of elution was detected when the digestive fluid from the lugworm (Arenicola marina), which contains serine peptidases, was subjected to affinity chromatography in a p-aminobenzamidine column. Chymotrypsin was not retained by the column, but trypsin was retained and was eluted by means of a competitive inhibitor (Eberhardt 1992). This can be explained by the stronger affinity of trypsins for benzamidine (Mares-Guia and Shaw 1965).
The theoretical molecular weight of lobster chymotrypsin (24 kDa) is similar to that of chymotrypsins from other decapods, such as the whiteleg shrimp (L. vannamei), which is 25 kDa (Van Wormhoudt et al. 1992), and the isochymotrypsins from the giant tiger prawn (P. monodon), kuruma shrimp (Marsupenaeus japonicus), and redtail prawn (Fenneropenaeus penicillatus) with molecular weights ranging from 25 to 28 kDa (Tsai et al. 1991); also, to that of other marine invertebrates, such as cuttlefish (Sepia officinalis) and lugworm (A. marina), with molecular weights of 28 and 25.4 kDa, respectively (Eberhardt 1992; Balti et al. 2012); and to those of Monterrey sardine (Sardinops sagax caerulea), 26 kDa (Castillo-Yáñez et al. 2006), and bovine chymotrypsin (B. taurus), 25 kDa (DelMar et al. 1979).
Under denaturing conditions, bovine chymotrypsin appears in SDS-PAGE as a protein composed of three polypeptide chains. Each polypeptide chain is produced during activation of chymotrypsinogen and held together by disulfide bridges (Neurath 1957). Our results showed that lobster chymotrypsin is composed of a single polypeptide chain because it appeared as a single band in electrophoresis after DTT/heat treatment (Fig. 2, lane 3). As other chymotrypsins in decapods, lobster chymotrypsin lacks four cysteine residues, which in bovine chymotrypsin form disulfide bonds that hold the peptide chains of the active form (Fig. 7, asterisk; Navarrete-del-Toro et al. 2015). To date, there is no experimental description of the zymogen activation process of chymotrypsin in decapods.
Even though trypsins and chymotrypsins share a similar folding structure, they have different primary specificities regarding differences in the S1 subsite (Perona and Craik 1997; Hedstrom 2002). Chymotrypsins hydrolyze peptide bonds from the carboxyl side with hydrophobic amino acid residues of Phe, Tyr, and Trp (Di Cera 2009), while trypsins hydrolyze peptide bonds formed by the positively charged polar amino acid residues Lys and Arg (Muhlia-Almazán et al. 2008). Lobster chymotrypsin hydrolyzed SAPNA but not BAPNA, most likely because it conserves the hydrophobic S1 pocket, as suggested by the primary structure (Fig. 7, bold).
Inhibition by PMSF indicates that the enzyme is a serine peptidase because PMSF is a highly electrophilic transition state analog that inactivates serine peptidases by reacting with the catalytic residue Ser195 (Hedstrom 2002). The purified enzyme has an optimal at pH 8.0 and is also able to carry out proteolysis at acid pH; this is not a common property of serine peptidases; therefore, the use of pepstatin A and E-64 helped to discard the possible contamination by aspartic and cysteine peptidases, which are active at acid pH. Finally, the enzyme was inhibited by chymostatin, to a lesser extent by TPCK and was not inhibited by TLCK. TLCK and TPCK are small molecules that bear a lysinyl and phenylalanyl moiety, respectively, while chymostatin is a peptide-derived aldehyde bearing a hydrophobic residue. Their inhibition potency is defined by the specificity of the peptidase. Considering this, TPCK and chymostatin inactivate chymotrypsins, while TLCK inactivates trypsins (Hedstrom 2002). Several reported chymotrypsins from marine invertebrates poorly react with TPCK (Tsai et al. 1991; Hernández-Cortés et al. 1997; Balti et al. 2012). Tsai et al. (1991) suggested that the interaction between giant tiger prawn chymotrypsin and the substrate involve extended subsites.
Kinetic parameters indicate that the lobster chymotrypsin has low affinity and is less efficient with the synthetic substrate SAPNA, compared with Monterrey sardine (Castillo-Yáñez et al. 2006) and bovine chymotrypsin (DelMar et al. 1979). However, low affinity towards the synthetic substrate is also a trait of other crustacean chymotrypsins (Tsai et al. 1991; Hernández-Cortés et al. 1997; Navarrete-del-Toro et al. 2015). Low affinity and efficiency at hydrolysis of SAPNA was associated with the lack of or low inhibitory effect of TPCK on chymotrypsins from decapods (Tsai et al. 1991). However, other chymotrypsins, show high affinity and efficiency towards the same substrate and is not inhibited by TPCK, such as chymotrypsin from cuttlefish (Balti et al. 2012). Analysis of the extended substrate binding sites of crustacean chymotrypsins could shed insight on the differences in inhibition profiles that they exhibit, in comparison with chymotrypsins from mammals.
Most chymotrypsins lose activity quickly at pH <5.0. After 1 h at pH 4.0, L. vannamei chymotrypsin loses 50 % of activity (Hernández-Cortés et al. 1997), S. sagax caerulea isochymotrypsins lose >95 % (Castillo-Yáñez et al. 2006; Castillo-Yañez et al. 2009), and S. officinalis chymotrypsin loses 70 % (Balti et al. 2012). Lobster chymotrypsin conserve up to 80 % activity after 1 h at pH 3.0; therefore, it is stable at mildly acid pH values. Even though the optimal pH for peptide bond hydrolysis of lobster chymotrypsin is within the alkaline pH, we demonstrated that the enzyme can hydrolyze protein at acid pH. Digestive peptidases active at acid pH have been reported in some marine invertebrates, but all of them belong to the aspartic and cysteine catalytic types (Teschke and Saborowski 2005; Rojo et al. 2010a, b). A recent report shows that digestive gland extracts from some crustacean decapods exhibit peptidase activity over a wide range of pH, possibly from serine peptidases (Navarrete del Toro et al. 2011). However, to the best of our knowledge, this is the first report of the purification and characterization of a serine peptidase bearing these characteristics.
Lobster chymotrypsin showed collagenase activity (Fig. 6b), and its primary structure shows the same residues forming the S1 binding pocket as the brachyurin from U. pugilator (Fig. 7, bold). Brachyurins are serine peptidases found in various species of crustaceans that exhibit mixed substrate specificity and marked collagenolytic activity (Tsai et al. 1986, 1991; Van Wormhoudt et al. 1992; Tsu et al. 1994; Rudenskaya 2003). The biological function of these enzymes is digestion of food protein that contain large quantities of collagen (Rudenskaya 2003). Collagenases of decapods are used in medicine, including treatment of burns, scars, and osteochondroses (Rudenskaya 2003). Collagenases are also important in collagen hydrolyzates for cosmetic application (Li et al. 2005) and in the food industry (Lima et al. 2009).
Temperature stability of proteins result from the number and strength of stabilizing intramolecular interactions, such as hydrogen bonds, hydrophobic interactions, salt bridges, and covalent interactions, such as disulfide bonds (D’Amico et al. 2002). Temperature stability of lobster chymotrypsin is similar to other marine chymotrypsins (Yang et al. 2009; Balti et al. 2012), although other representatives exhibit higher thermal stability (Hernández-Cortés et al. 1997).
Organic solvents are important components in different industrial enzymatic processes; for instance, those requiring solubilization of non-polar substrates and decrease of water-dependent side reactions. Also, peptidases stable in organic solvents can be used as biocatalysts for peptide synthesis. However, peptidases in general are inactivated and show low reaction rates under such conditions (Gupta 1992). Most solvent-tolerant peptidases that have been studied come from microorganisms that thrive in the presence of high concentrations of organic solvents (Geok et al. 2003). Only a few reports mention solvent-tolerant proteases from decapods (Saborowski et al. 2004). Lobster chymotrypsin was also affected to varying degrees by different solvents. Even though DMSO and toluene markedly reduced the activity of lobster chymotrypsin, it is also remarkable that lobster chymotrypsin retains >80 % of its activity when tested in the presence of acetone, ethanol, propanol, and butanol.
Our data confirms that the purified enzyme from the gastric juice of California spiny lobster (P. interruptus) is a chymotrypsin-like serine peptidase. Lobster chymotrypsin showed collagenolytic activity, but further investigation is needed to explore this characteristic and substrate specificity. Lobster chymotrypsin exhibited particular biochemical characteristics: highly stable at acid pH and able to hydrolyze protein within a wide range of pH from its optimal alkaline pH and even at pH 3.0. Thus, lobster chymotrypsin is responsible, to some extent, for the previously reported proteolytic activity at acidic pH. We also showed a high retention of activity when the chymotrypsin was incubated with different solvents. Considering that resistance to harsh conditions are characteristics of enzymes that biotechnologists constantly intend to use in new processes, the particularities of lobster chymotrypsin could have advantages in biotechnological applications and increase the economic value of this marine resource.
References
Balti R, Bougherra F, Bougatef A, Hayet BK, Nedjar-Arroume N, Dhulster P, Guillochon D, Nasri M (2012) Chymotrypsin from the hepatopancreas of cuttlefish (Sepia officinalis) with high activity in the hydrolysis of long chain peptide substrates: purification and biochemical characterisation. Food Chem 130:475–484
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Castillo-Yañez FJ, Pacheco-Aguilar R, Garcia-Carreño FL, Navarrete-Del Toro MDLA (2004) Characterization of acidic proteolytic enzymes from Monterey sardine (Sardinops sagax caerulea) viscera. Food Chem 85:343–350
Castillo-Yáñez FJ, Pacheco-Aguilar R, García-Carreño FL, Navarrete-Del Toro MA, Félix López M (2006) Purification and biochemical characterization of chymotrypsin from the viscera of Monterey sardine (Sardinops sagax caeruleus). Food Chem 99:252–259
Castillo-Yañez FJ, Pacheco-Aguilar R, Lugo-Sanchez ME, Garcia-Sanchez G, Quintero Reyes IE (2009) Biochemical characterization of an isoform of chymotrypsin from the viscera of Monterey sardine (Sardinops sagax caerulea), and comparison with bovine chymotrypsin. Food Chem 112:634–639
Celis-Guerrero LE, García-Carreño FL, Navarrete Del Toro MA (2004) Characterization of proteases in the digestive system of spiny lobster (Panulirus interruptus). Mar Biotechnol 6:262–269
D’Amico S, Claverie P, Collins T, Georlette D, Gratia E, Hoyoux A, Meuwis M-A, Feller G, Gerday C (2002) Molecular basis of cold adaptation. Philos Trans R Soc Lond B Biol Sci 357:917–925
DelMar EG, Largman C, Brodrick JW, Geokas MC (1979) A sensitive new substrate for chymotrypsin. Anal Biochem 99:316–320
Di Cera E (2009) Serine proteases. IUBMB Life 61:510–515
Díaz-Tenorio LM, García-Carreño FL, Navarrete del Toro MA (2006) Characterization and comparison of digestive proteinases of the Cortez swimming crab, Callinectes bellicosus, and the arched swimming crab, Callinectes arcuatus. Invertebr Biol 125:125–135
Eberhardt J (1992) Isolation and characterization of 5 serine proteases with trypsin-like, chymotrypsin-like and elastase-like characteristics from the gut of the lugworm Arenicola marina (L.) (Polychaeta). J Comp Physiol B 162:159–167
El Hadj AN, Hmidet N, Zouari-Fakhfakh N, Ben Khaled H, Nasri M (2010) Alkaline chymotrypsin from striped seabream (Lithognathus mormyrus) viscera: purification and characterization. J Agric Food Chem 58:9787–9792
Fong WP, Chan EY, Lau KK (1998) Isolation of two chymotrypsins from grass carp. Biochem Mol Biol Int 45:409–418
García-Carreño F, Dimes L, Haard N (1993) Substrate-gel electrophoresis for composition of molecular weight of proteinases or protteinaceous proteinase inhibitors. Anal Biochem 214:65–69
Geok LP, Razak CNA, Abd Rahman RNZ, Basri M, Salleh AB (2003) Isolation and screening of an extracellular organic solvent-tolerant protease producer. Biochem Eng J 13:73–77
Gráf L, Szilágyi L, Venekei I (2013) Chymotrypsin. In: Rawlings ND, Salvesen G (eds) Handbook of Proteolytic Enzymes, 3rd edn. Academic Press, San Diego, p 2626–2633
Gupta MN (1992) Enzyme function in organic solvents. Eur J Biochem 203:25–32
Hedstrom L (2002) Serine protease mechanism and specificity. Chem Rev 102:4501–4523
Hernández-Cortés P, Whitaker JR, García-Carreño F (1997) Purification and characterization of chymotrypsin from Penaeus vannamei. J Food Biochem 21:497–514
Heu MS, Kim HR, Pyeun JH (1995) Comparison of trypsin and chymotrypsin from the viscera of anchovy, Engraulis japonica. Comp Biochem Physiol B: Biochem Mol Biol 112:557–567
Klomklao S (2008) Digestive proteinases from marine organisms and their applications. Songklanakarin J Sci Technol 30:37–46
Kristjansson MM (1991) Purification and characterization of trypsin from the pyloric ceca of rainbow trout (Oncorhynchus mykiss). J Agric Food Chem 39:1738–1742
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Le Chevalier P, Sellos D, Van Wormhoudt A (1995) Purification and partial characterization of chymotrypsin-like proteases from the digestive gland of the scallop Pecten maximus. Comp Biochem Physiol B Biochem Mol Biol 110:777–784
Li GY, Pukunaga S, Takenouchi K, Nakamura F (2005) Comparative study of the physiological properties of collagen, gelatin and collagen hydrolysate as cosmetic materials. Int J Cosmet Sci 27:101–106
Lima CA, Rodrigues PMB, Porto TS, Viana DA, Lima Fihlo JL, Porto ALF, Carneiro da Cunha MG (2009) Production of a collagenase from Candida albicans URM3622. Biochem Eng J 43:315–320
Mares-Guia M, Shaw E (1965) Studies on the Active Center of Trypsin: the binding of amidines and guanidines as models of the substrate side chain. J Biol Chem 240:1579–1585
Muhlia-Almazán A, García-Carreño FL (2002) Influence of molting and starvation on the synthesis of proteolytic enzymes in the midgut gland of the white shrimp Penaeus vannamei. Comp Biochem Physiol B Biochem Mol Biol 133:383–394
Muhlia-Almazán A, Sánchez-Paz A, García-Carreño FL (2008) Invertebrate trypsins: a review. J Comp Physiol B 178:655–672
Navarrete del Toro MDLA, García-Carreño F, López MD, Celis-Guerrero L, Saborowski R (2006) Aspartic proteinases in the digestive tract of marine decapod crustaceans. J Exp Zool A Comp Exp Biol 305:645–654
Navarrete del Toro MA, García-Carreño FL, Córdova-Murueta JH (2011) Comparison of digestive proteinases in three penaeids. Aquaculture 317:99–106
Navarrete-del-Toro MA, García-Carreño Fernando L, Hernández-Cortés P, Molnár T, Gráf L (2015) Biochemical characterisation of chymotrypsin from the midgut gland of yellowleg shrimp, Penaeus Californiensis. Food Chem 173:147–155
Neurath H (1957) The activation of zymogens. In: Anfisen CB, Anson ML, Bailey K, Edsall JT (eds) Advances in Protein Chemistry. Academic Press, New York, p 319–386
Perera E, Fraga I, Carrillo O, Díaz-Iglesias E, Cruz R, Báez M, Galich GS (2005) Evaluation of practical diets for the Caribbean spiny lobster Panulirus argus (Latreille, 1804): effects of protein sources on substrate metabolism and digestive proteases. Aquaculture 244:251–262
Perera E, Moyano FJ, Díaz M, Perdomo-Morales R, Montero-Alejo V, Carrillo O, Galich GS (2008a) Polymorphism and partial characterization of digestive enzymes in the spiny lobster Panulirus argus. Comp Biochem Physiol B Biochem Mol Biol 150:247–254
Perera E, Moyano FJ, Díaz M, Perdomo-Morales R, Montero-Alejo V, Rodriguez-Viera L, Alonso E, Carrillo O, Galich GS (2008b) Changes in digestive enzymes through developmental and molt stages in the spiny lobster, Panulirus argus. Comp Biochem Physiol B Biochem Mol Biol 151:250–256
Perona JJ, Craik CS (1995) Structural basis of substrate specificity in the serine proteases. Protein Sci 4:337–360
Perona JJ, Craik CS (1997) Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J Biol Chem 272:29987–29990
Raae AJ, Walther BT (1989) Purification and characterization of chymotrypsin, trypsin and elastase like proteinases from cod (Gadus morhua L.). Comp Biochem Physiol B 93:317–324
Rojo L, Muhlia-Almazan A, Saborowski R, García-Carreño F (2010a) Aspartic cathepsin D endopeptidase contributes to extracellular digestion in clawed lobsters Homarus americanus and Homarus gammarus. Mar Biotechnol 12:696–707
Rojo L, Sotelo-Mundo R, García-Carreño F, Gráf L (2010b) Isolation, biochemical characterization, and molecular modeling of American lobster digestive cathepsin D1. Comp Biochem Physiol B Biochem Mol Biol 157:394–400
Roy P, Colas B, Durand P (1996) Purification, kinetical and molecular characterizations of a serine collagenolytic protease from greenshore crab (Carcinus maenas) digestive gland. Comp Biochem Physiol B Biochem Mol Biol 115:87–95
Rudenskaya GN (2003) Brachyurins, serine collagenolytic enzymes from crabs. Russ J Bioorg Chem 29:101–111
Saborowski R, Sahling G, Navarrete del Toro MA, Walter I, García-Carreño FL (2004) Stability and effects of organic solvents on endopeptidases from the gastric fluid of the marine crab Cancer pagurus. J Mol Catal B Enzym 30:109–118
Shi XZ, Zhao XF, Wang JX (2008) Molecular cloning and expression analysis of chymotrypsin-like serine protease from the Chinese shrimp, Fenneropenaeus chinensis. Fish Shellfish Immunol 25:589–597
Stauffer C (1989) Enzyme assays for food scientist. Chapman and Hall, New York, p 61–76
Teschke M, Saborowski R (2005) Cysteine proteinases substitute for serine proteinases in the midgut glands of Crangon crangon and Crangon allmani (Decapoda: Caridea). J Exp Mar Biol Ecol 316:213–229
Tsai I-H, Chuano K-L, Chuang JL (1986) Chymotrypsins in digestive tracts of crustacean decapods (Shrimps). Comp Biochem Physiol B 85:235–239
Tsai IH, Lu PJ, Chuang JL (1991) The midgut chymotrypsins of shrimps (Penaeus monodon, Penaeus japonicus and Penaeus penicillatus). Biochim Biophys Acta 1080:59–67
Tsu CA, Perona JJ, Schellenberger V, Turck CW, Craik CS (1994) The substrate specificity of Uca pugilator collagenolytic serine protease 1 correlates with the bovine type I collagen cleavage sites. J Biol Chem 269:19565–19572
Van Wormhoudt A, Le Chevalier P, Sellos D (1992) Purification, biochemical characterization and N-terminal sequence of a serine-protease with chymotrypsic and collagenolytic activities in a tropical shrimp, Penaeus vannamei (Crustacea, Decapoda). Comp Biochem Physiol B 103:675–680
Yang F, Su W-J, Lu B-J, Wu T, Sun L-C, Hara K, Cao M-J (2009) Purification and characterization of chymotrypsins from the hepatopancreas of crucian carp (Carassius auratus). Food Chem 116:860–866
Acknowledgments
We thank Patricia Hernandez-Cortes for technical assistance and Ira Fogel for comprehensive editing services (CIBNOR). This study was supported by Consejo Nacional de Ciencia y Tecnología (CONACYT grant 80935 to F.G.C.). B.B.V. is a recipient of a graduate fellowship (CONACYT 277859). We thank Dr. Reinhard Saborowski at AWI, Germany, for supporting this research.
Conflict of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bibo-Verdugo, B., Rojo-Arreola, L., Navarrete-del-Toro, M.A. et al. A chymotrypsin from the Digestive Tract of California Spiny Lobster, Panulirus interruptus: Purification and Biochemical Characterization. Mar Biotechnol 17, 416–427 (2015). https://doi.org/10.1007/s10126-015-9626-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-015-9626-z