
Abstract
The microbiome is an important consideration for the conservation of endangered species. Studies provided evidence of the effect of behavior and habitat change on the microbiota of wild animals and reported various inferences. It indicates the complexity of factors influencing microbiota diversity, including incomplete sampling procedures. Data abnormality may arise due to the procedures warranting preliminary analysis, such as rarefaction, before downstream analysis. This present study demonstrated the effect of data rarefaction and aggregation on the comparison of wild rusa deer’s gut microbial diversity. Eighty-five feces samples were collected from 11 deer populations inhabiting three national parks in Java and Bali islands. Using the Illumina Nova-Seq platform, fragments of 16s rRNA gene were sequenced, and raw data of 51,389 reads corresponding to 2 domains, 22 phyla, 45 classes, 83 orders, 182 families, and 460 genera of bacteria were obtained. Data rarefaction was applied at two different library sizes (minimum and fixed) and aggregation (11 populations into 3 research sites) to investigate its effect on the microbial diversity comparison. There are significant differences in alpha diversity between populations, but not research sites, at all library sizes of rarefaction. A similar finding is also found in beta diversity. Moreover, data rarefaction and aggregation result in different values of the diversity metrics. This present study shows that statistical analysis remains a substantial concern in microbiome studies applied to conservation biology. It suggests reporting a more detailed data normalization in microbiome studies as an inherent control of suboptimal sampling, particularly when involving feces.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The microbiome study of wild animals has received much attention in conservation biology. In that field, interactions of animal hosts and microbiota are an essential aspect that should be considered for conservation measures of endangered species (Bahrndorff et al. 2016; Trevelline et al. 2019; Wei et al. 2019; Zhu et al. 2021). Several recent studies provide evidence of the effect of behavioral adaptation on the abundance of the microbiota of wild animals. For example, Y. Sun et al. (2020) reported that Firmicutes-rich gut microbiota has a beneficial impact on the energy intake of the wild Alpine musk deer (Moschus chrysogaster). Tang et al. (2020) stated that a diverse microbiota in semi-wild Przewalski’s horses (Equus ferus przewalskii) provided a higher metabolic potential to utilize the complex plants. Yang et al. (2020) suggested adjusting the gut microbial community through diet training before individual release to increase the survival rates of Yangtze sturgeon (Acipenser dabryanus). Meanwhile, other studies showed the effect of habitat change and diet availability on the microbiota of wild animals. For example, Barelli et al. (2015) reported the importance of an undisturbed forest patch for securing healthy microbiota of the Udzungwa red colobus monkey (Procolobus gordonorum), an endangered primate species. Additionally, Murray et al. (2020) discovered that changes in the habitat and the diet of birds modify gut microbial composition influencing health and pathogen susceptibility. The effect is particularly pronounced on the microbiota diversity because environmental perturbations most likely affect alpha diversity. Therefore, comparing this diversity represents an initial analysis of many microbiome studies (Willis 2019).
Particularly for diversity comparison, several studies investigated the gut microbiota in both wild and captive environment settings and indicated data analysis issues. For illustration, research shows that comparing microbiota diversities of captive and wild animals such as black rhinos (Gibson et al. 2019), Pere David’s deer (Sun et al. 2019), gaurs and mithuns (Prabhu et al. 2020), giant pandas (Guo et al. 2019), and other 24 vertebrate species (Alberdi et al. 2021) results in various conclusions. The variability of the results indicates the complexity of factors influencing the microbiota diversity of wild animals. Despite the natural variation, the sampling procedures and data analysis could also contribute to the disparity of the results. Particularly for wild animals, the collection of degraded and contaminated samples is inevitable. This practice increases the risk of suboptimal sampling procedures, including a low PCR efficacy, resulting in suboptimal microbiota sequencing. This suggests that statistical analysis remains a substantial concern in conservation biology studies to decrease the impact of such artefactual factors (Zhu et al. 2021).
A crucial issue in the statistical analysis of high-throughput microbiota sequencing is the data sparsity and high variation of sequence number (Tsilimigras and Fodor 2016; Pan 2021), which are typically overcome through the application of normalization. The sparsity occurs naturally in the microbiota data and is usually indicated by the absence or very low abundance of many taxa in the samples, in addition to the high abundance of several others. The problem of high variation in the number of sequences, caused by confounding factors of the natural characteristic of the microbiota, sampling procedures, and sequencing technique (Paulson et al. 2013), increases the complexity of the data analysis. These issues pose a problem in statistical modeling as both parametric and nonparametric models are unsuitable for analyzing data with too many zeros (Xia et al. 2018). Therefore, ignoring them may result in biased parameter estimation and false inference in the downstream data analysis. Normalization is a technique to handle such problems. It is a process in which the data is transformed to exclude artifactual biases in the original measurement (Weiss et al. 2017).
Furthermore, scaling methods (Paulson et al. 2013), rarefaction (Hughes and Hellmann 2005), and log-ratio transformation (Aitchison 1982) are among the popular normalization techniques. There is no consensus on which method is the most appropriate for all cases, but it depends on the data characteristics and objective of the study, including the magnitude of data variation (i.e., average library size; Weiss et al. (2017)), ecological consideration of original microbiota (McKnight et al. 2019), the importance of phylogenetic tree in the analysis (Liu et al. 2020), and the importance of taxa abundance in the study (Lin and Das 2020). The first two articles suggested the superiority of rarefaction over other methods when microbiota diversity is of concern. Meanwhile, the two last proposed log-ratio transformations when the difference in microbiota abundance is the focus of the study. The normalization process is thorough, involves many parameters, and is infrequently reported in detail.
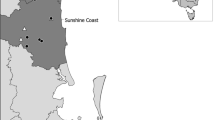
In the present study, data normalization (rarefaction technique) was applied to the gut bacterial diversity of wild rusa deer (Cervus timorensis) comparison to demonstrate its importance using empirical data from the field. The deer is a protected species in Indonesia that natively inhabits Java and Bali islands. At this location, the deer are distributed unevenly in several conservation areas, including savannas in the Baluran, Bali Barat, and Alas Purwo national parks. Baluran and Bali Barat national parks are distanced by approximately 30 km and spaced by a strait. Meanwhile, the distance between Baluran and Alas Purwo national parks is approximately 90 km. Each ecosystem of the savannas is unique in terms of vegetation and abiotic environment. Arundinella setosa, Desmodium laxiflorum, Chromolaena odorata, and Flacourtia rukam dominated vegetation in the savanna of Alas Purwo National Park. The savanna of Bali Barat National Park is dominated by Calamagrostis australis and Borassus flabellifer, while Baluran National Park primarily consists of Desmodium laxiflorum, Azadirachta indica, Polytrias indica, Dichanthium caricosum, Ziziphus mauritiana, and Acacia nilotica (Sutomo and van Etten 2021). The savanna of Bali Barat is significantly richer in species than Baluran and Alas Purwo national parks. Variations in precipitation, temperature, altitude, fire regimes, and human intervention are likely the potential drivers of the uniqueness of the savanna (Whitten et al. 1996). It has been hypothesized that these habitat variations cause fluctuations in food availability and diet, leading to differences in the microbiota diversity of the deer. However, in addition to natural variation, suboptimal sample collection and sequencing procedures due to non-invasive sampling may also affect the comparison. Our study aims to compare the bacterial diversity of several deer populations residing in three national parks on the Java and Bali islands. Through this comparison, we envisage a better understanding of the effects of data normalization and aggregation on the results.
Methods
Data collection started with gathering feces of wild rusa deer populations, followed by genomic DNA isolation, high-throughput sequencing, quality filtering, and annotating of data. Feces samples were collected in the savannas of three national parks (research sites), Baluran, Bali Barat, and Alas Purwo, located in the eastern part of Java Island and Bali Island, as shown in Fig. 1. The recent distribution of the deer populations was surveyed within those savannas before feces collection. In pre-determined populations, a series of circular plots with a 1 m radius were laid every 5 m along a line transect approximately 10 m apart, covering the area. Furthermore, fresh feces (indicated by moist or shiny surface) with a similar appearance were collected from each circular plot. Ten to twenty pellets of the deer feces were preserved in 96% ethanol inside 50-mL conical tubes. They were collected early morning (06.00–08.00) to minimize DNA degradation from sun exposure. The DNA was isolated from the feces within 2 weeks of collection using a DNeasy PowerSoil Pro® kit (Qiagen, Hilden, Germany). The extraction was performed following the whole manufacture protocol except overnight cell lysis.

Map showing research sites and populations where feces samples of the rusa deer were collected: Baluran (1, Bama; 2, Bekol; 3, Bilik; 4, Merak; 5, Datuk populations), Bali Barat (6, Brumbun; 7, Pura Segara; 8, Prapat Agung; 9, Octagon populations), and Alas Purwo (10, Plengkung; 11, Sadengan populations) national parks
Furthermore, the extracted DNA was quantified using a Quantus® Fluorometer (Promega Inc.) and processed further for high-throughput sequencing. Before the sequencing, the feces’ host was confirmed by PCR amplification using specific primers and Sanger sequencing of subsamples (Iman et al. 2024). For high-throughput sequencing, the V3-V4 regions from the 16s rRNA gene were amplified by PCR using a primer couple originally developed by Klindworth et al. (2013). PCR conditions were as follows: initial denaturation (98 °C, 3 min), followed by 38 cycles of denaturation (98 °C, 30 s), annealing (55 °C, 45 s), extension (72 °C, 60 s), and final extension (72 °C, 10 min). This PCR was followed by a PCR clean-up using HighPrep PCR magnetic beads (MagBio Genomics, Gaithersburg, MD) following the manufacturer’s protocol. Subsequently, an index PCR with Nextera tags allowed us to link each sequence to its corresponding sample and a second PCR clean-up followed. Resulting libraries were then quantified using Quant-iT™ PicoGreen™ on a plate reader following the manufacturer’s instructions. Libraries were normalized to 5 nM and were finally pooled together and sent to the GIGA sequencing platform (University of Liège) which performed the sequencing on a Nova-Seq 6000 (Illumina) using a 2 × 250 bp kit. The raw sequences were treated using a modified version of the bioinformatical script found in André et al.’s (2017) study, consisting of both FASTX-Toolkit (Hannon 2010) and USEARCH (Edgar 2010) functions. The clustering was performed using the swarm function (Mahé et al. 2021) with default parameters. After the filtering, the data was annotated using the RDP classifier function (Wang et al. 2007) and its corresponding database (v 2.13) using a 0.8 cutoff value.
Following the annotation, further downstream analysis was performed to investigate the influence of data normalization on diversity comparison. Samples that had less than 100 reads were excluded from the analysis. To mitigate the issue of incomplete sampling procedures, the occurrence of high variation in sequence numbers of each operational taxonomic unit (OTU) was evaluated, and the data were normalized. The evaluation was primarily based on the difference in abundance of each OTU among the samples, as suggested by Weiss et al. (2017). The significance of the difference was tested using one-way ANOVA. Data normalization was performed using the rarefaction technique at a minimum library size and a fixed (500) sequence read. This fixed read was chosen because it captured the most taxonomic tags, although eliminating samples with reads less than 500. Normalized and unnormalized data were analyzed further by observing the differences in alpha and beta diversities. Alpha diversity was evaluated through six metrics, including Observed, Chao1, ACE, Shannon’s, Simpson’s, and Fisher’s diversity indices, and analyzed with the ANOVA test. Moreover, beta diversity was assessed using ordination analyses of principal coordinate analysis (PCoA) and non-metric multidimensional scaling (NMDS) based on a Bray-Curtis dissimilarity matrix. Diversity comparisons were conducted at the genus level among populations and aggregated populations (national parks). Statistical tests were performed under an R-statistic environment either independently or embedded into a web-based application (MicrobiomeAnalyst; Chong et al. (2020)).
Result
Bacterial DNA at 16S rRNA gene was successfully sequenced and identified from the 85 deer feces collected from populations in Baluran (Bama, n = 4; Bekol, n = 4; Bilik, n = 2; Datuk, n = 2; Merak, n = 6), Bali Barat (Brumbun, n = 2; Octagon, n = 6; Prapat Agung, n = 6; Pura Segara, n = 7), and Alas Purwo (Plengkung, n = 10; Sadengan, n = 14) national parks. The raw data of 51,389 reads corresponding to 2 domains, 22 phyla, 45 classes, 83 orders, 182 families, and 460 genera of bacteria were obtained. Removing extremely low-depth sequences (< 100 reads) did not affect the statistics but eliminated 32 reads and a whole data set of a feces sample. The data showed a sparsity of 0.83 and singletons of 105 (22.8%) reads. Considering these values, there is a concern about data sparsity and high variation in sequence numbers. Initial observation indicates a wide variety of library sizes per sample, ranging from 338 to 1345 reads.
Further analysis presents a significant difference in the abundance of each OTU among the samples (ANOVA test; p < 0.05). This showed a probability of incomplete sampling procedures and suggested data normalization. Rarefaction at minimum library size (338 reads) preserved the sample size (84 feces samples) but reduced the total number of sequences (28,392 reads) and corresponding OTU (295 genera). Meanwhile, at a maximum of 500 sequence reads per sample, it resulted in the highest taxonomic tags at the expense of a substantial decrease in sequence reads and the removal of 22 samples. Subsequently, there were 31,500 sequence reads corresponding to 367 genera.
Except for the Simpson Diversity Index, there is a significant difference in alpha diversity among populations as measured by all metrics (p < 0.05; Fig. 2). Consistent conclusions were obtained from the three data sets: such as no rarefaction, rarefied at 338, and 500 sequences read/samples. The rarefactions tend to decrease the values of richness-influenced indices according to the number of OTUs and narrow the data distribution (except the Simpson Index), as presented in Fig. 2. In that figure, it was noticed that the Prapat Agung population showed the highest diversity index and variations in all metrics except the Simpson Index. However, it did not affect the statistical inferences of the comparisons.

Boxplots showing the comparison of bacterial diversity by six metrics among 11 populations of rusa deer at three rarefactions: a no rarefaction, b rarefaction at minimum library size (338 reads), and c rarefaction at fixed library size (500 reads). The p-value of the ANOVA test is provided below the plot
Different inferences were found when aggregating the samples into each research site of origin (national park) and evaluating their diversity. There was no significant difference in the alpha diversity of bacteria from the Baluran, Bali Barat, and Alas Purwo national parks (p > 0.05; Fig. 3). Similar to the previous result, the rarefactions tend to decrease indices. However, a clear pattern of the association between data distribution and rarefactions was not observed.

Boxplots showing the comparison of rusa deer’s bacterial diversity on six metrics at three research sites and rarefactions: a no rarefaction, b rarefaction at minimum library size (338 reads), and c rarefaction at fixed library size (500 reads). The p-value of the ANOVA test is provided below the plot
The beta diversity analysis showed distinctive ordinations of bacterial communities from 11 rusa deer populations. Furthermore, PCoA and NMDS analysis showed significant differences (p < 0.05) with some overlap (R2 = 0.39–0.46 for PCoA and R = 0.43–0.60 for NMDS). The application of rarefactions did not affect these results but changed the values of both R- and R2-statistics and variants of dissimilarity as shown by the elliptical area in Fig. 4. Rarefaction at a fixed library size (500 reads) showed a consistent result in both PCoA and NMDS analysis. The result revealed that Prapat Agung has the largest variance of dissimilarity, followed by Sadengan and Pura Segara populations. It corresponded to alpha diversity metrics as shown by the length of boxplots in Fig. 2c. Meanwhile, other rarefaction applications did not yield consistent variance of beta diversity. It indicated that the rarefaction at fixed library size effectively removes bias introduced by sampling and sequencing techniques and reflects better the nature of microbiota variation.

Chart showing ordinations of rusa deer’s bacterial diversity using PCoA (left) and NMDS (right) techniques among 11 populations at three rarefactions: a no rarefaction, b rarefaction at minimum library size (338 reads), and c rarefaction at fixed library size (500 reads). Ellipses represented a 95% confidence limit. The p-value of the test is provided below the plot
The group of bacterial communities was also significantly identified when the populations were aggregated into three research sites. Despite the R-value being smaller than among the population level of analysis, PCoA and NMDS analysis showed distinctive ordinations (p < 0.05; Fig. 5). Furthermore, unlike the population level of analysis, this research sites analysis showed insensitiveness of the rarefaction and ordination techniques to the variance of dissimilarity. Bali Barat constantly has the largest variance followed by Baluran and Alas Purwo.

Chart showing ordinations of rusa deer’s bacterial diversity using PCoA (left) and NMDS (right) techniques between three research sites at three rarefactions: a no rarefaction, b rarefaction at minimum library size (338 reads), and c rarefaction at fixed library size (500 reads). The p-value of the test is provided below the plot
Discussion
Interest in rarefaction analyses, sample aggregation, and diversity comparisons
This study provides evidence of a significant difference in alpha and beta diversity comparisons in most sample diversity metrics and aggregations, disregarding the rarefaction applications. There is a possibility that when the natural variation of bacterial community is high, as in this case, rarefaction does not affect the comparisons. It aligns with Hong et al. (2022) notion that rarefaction might not affect statistical inference much as long as the underlying biological variation in microbial composition is large enough among samples. Furthermore, the metrics commonly used for measuring alpha diversity behave similarly across rarefactions and sample aggregations, except for the Simpson Index. We provide evidence that diversity indices tend to decrease as the number of OTUs diminishes because of rarefactions. The number of OTUs is directly proportional to richness which strongly influences the diversity index. It was observed that the Simpson Index has no significant difference in bacterial diversity populations or sites. Due to sensitivity to evenness rather than richness and insensitivity to the contribution of rare taxa (DeJong 1975), the insignificant differences in the index indicate that all data sets similarly have several taxa with high abundance. Additionally, the beta diversity analysis shows consistent significant ordination regardless of adjustments in rarefactions and sample aggregations. This result is identical to previous research evidencing the insensitiveness of beta diversity to environmental variations (Guo et al. 2019; Sun et al. 2019; Gibson et al. 2019; Prabhu et al. 2020). It is worth noting that although rarefaction applications do not affect inferences, they influence data variance.
Unlike beta, alpha diversity of microbiota is considered to be sensitive to environmental variation thus emphasizing the importance of sample representativeness. Our results show an insignificant difference in alpha, but not beta, diversity when aggregating 11 samples into 3 research sites. The aggregation weakens the variation of bacterial diversity. The rusa deer gut microbiota is supposed to reflect the population’s microhabitat conditions. Therefore, aggregating samples at the landscape level (research sites) may not appropriately represent natural differences and mask the high bacterial community variation among populations. It illustrates the concern of the unrepresentativeness of sampling. The same concern, but in a different situation, is also shown in the results of several studies reporting insignificant alpha diversity when comparing the microbiota of wild and captive animals. Gibson et al. (2019) compared the diversity of captive (n = 17) and wild populations (n = 8; opportunistically sampled from the wild) of black rhinos and found no significant difference in alpha diversity. A similar result was obtained from the wild (n = 10) and captive gaur (n = 10) populations (Prabhu et al. 2020). Additionally, involving six samples of wild and seven samples of captive Père David’s deer, C. Sun et al. (2019) also reported a non-significant difference in alpha diversity of the deer’s microbiota. Likely, the small sample size may not adequately represent the microbiota diversity of the animals living in those microhabitats, as proposed by West et al. (2019). Meanwhile, involving a large and widely distributed sample size (wild = 81 samples; captive = 49 samples), Guo et al. (2019) reported a significant difference in alpha diversity in the microbiota of giant pandas. The discrepancy suggests the importance of matching the variation between microbiota and habitat when conducting alpha diversity comparisons.
An impact of habitats on the deer microbiota
Appropriate data analysis results in a better understanding of the interactions between the habitat of wild animals, gut microbiota community, and fitness which is important for conservation biology. In this present study, it was discovered that alpha and beta diversities were significantly different among populations. Variation in the ecological condition of the deer habitats causes differences in its microbiota community. At the research site level (among national parks), a positive association between the variation of habitat and gut bacteria of rusa deer was speculated after rarefactions at a fixed library size. Sutomo and van Etten (2021) reported that the diversity index of Shannon-Wiener of plant species in the savanna of Alas Purwo, Bali Barat, and Baluran national parks was 2.6, 5.0, and 3.10 respectively, while the Shannon Index of deer’s microbiota was 2.49, 2.55, and 2.51 (Fig. 3c). Although association analysis at this level is not robust, however, this pattern advises further studies on the association between microbiota and the habitat of the deer. Additionally, it provides preliminary evidence of the effectiveness of data normalization in revealing true interactions. Several studies have reported a possible association between habitat and microbiota diversity of wildlife. Amato et al. (2013) reported an association between the composition of the gut microbiota and habitat variation of the black howler monkey in a degraded forest. A similar finding was also reported in the red colobus monkey (Barelli et al. 2015). Besides, in the wild-captive habitat setting, Borbón-García et al. (2017) showed a correlation between the decreased microbiota diversity of Andean bears and captive habitat conditions, particularly due to a low variation of diet. However, the possibility of incomplete sampling procedures and the sample size is rarely discussed as an important contributor to the variation of the result. It hinders the disentanglement of the effect of natural differences and sampling procedures on the statistical inferences from comparing microbiota diversity. Finally, any result divergence from the understanding prompts an evaluation of sampling procedures and an explanation.
Moreover, our study supports Zhu et al. (2021), which emphasizes the importance of in-depth data analysis for the application of microbiome study in conservation biology. In this field, there is a need to implement non-invasive genetic sampling for wild animal studies (Zemanova 2019). However, as the scheme unavoidably involves degraded samples, particularly in tropical areas (Goossens and Salgado-lynn 2013), the implementation should consider the involvement of field control samples as recommended by Cando-Dumancela et al. (2021) and be complemented by comprehensive data analysis to avoid biased inferences. The control samples may include soil near the place where the samples were collected. This would allow the separation of the true microbiota from the environmental bacterial contaminations. Moreover, specific bacterial groups are only found in the intestines and not in the environment. Especially when studying the microbiota of wild animals using high-throughput sequencing, there is a risk that difference in sample quality causes a large variation in microbes that can be sequenced. It results in data sparsity and high variation of library size between samples, suggesting normalization before any statistical analysis. Despite not significantly affecting the comparison inference, rarefactions alter the measurement and coefficient determination (R2) of alpha and beta diversities, respectively. We suggest that data normalization has to be reported in more detail in the microbiome study of wild animals as an inherent control of suboptimal sampling, particularly when using non-invasive samples. This would ensure that published data has been evaluated for comprehensive analysis, including the possibility of biased inference due to incomplete sampling.
Conclusions
The alpha diversity of the microbiota exhibits significant differences across 11 populations of rusa deer, but not research sites, throughout all library sizes of rarefaction. This tendency is similarly reflected in beta diversity. Furthermore, data rarefaction and aggregation result in different values of the diversity metrics. As a consequence, it is imperative to accurately normalize the data for meaningful comparisons of microbiota diversity, especially when dealing with wild animals and an incomplete sampling approach. We highly recommend conducting a thorough analysis of microbiota data and careful data normalization before downstream statistical analysis to avoid spurious outcomes.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Aitchison J (1982) The statistical analysis of compositional data. J R Statist Soc B 44(2):139–177. https://doi.org/10.1111/j.2517-6161.1982.tb01195.x
Alberdi A, Martin Bideguren G, Aizpurua O (2021) Diversity and compositional changes in the gut microbiota of wild and captive vertebrates: a meta-analysis. Sci Rep 11:22660. https://doi.org/10.1038/s41598-021-02015-6
Amato KR, Yeoman CJ, Kent A, Righini N, Carbonero F, Estrada A, Rex Gaskins H, Stumpf RM, Yildirim S, Torralba M, Gillis M, Wilson BA, Nelson KE, White BA, Leigh SR (2013) Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J 7:1344–1353. https://doi.org/10.1038/ismej.2013.16
André A, Mouton A, Millien V, Michaux J (2017) Liver microbiome of Peromyscus leucopus, a key reservoir host species for emerging infectious diseases in North America. Infect Genet Evol 52:10–18
Bahrndorff S, Alemu T, Alemneh T, Lund Nielsen J (2016) The microbiome of animals: implications for conservation biology. Int J Genomics 2016:1–7. https://doi.org/10.1155/2016/5304028
Barelli C, Albanese D, Donati C, Pindo M, Dallago C, Rovero F, Cavalieri D, Michael TK, Christine HH, De Filippo C (2015) Habitat fragmentation is associated to gut microbiota diversity of an endangered primate: implications for conservation. Sci Rep 5:14862. https://doi.org/10.1038/srep14862
Borbón-García A, Reyes A, Vives-Flórez M, Caballero S (2017) Captivity shapes the gut microbiota of Andean bears: insights into health surveillance. Front Microbiol 8:1316. https://doi.org/10.3389/fmicb.2017.01316
Cando-Dumancela C, Liddicoat C, McLeod D, Young JM, Breed MF (2021) A guide to minimize contamination issues in microbiome restoration studies. Restor Ecol 29:e13358. https://doi.org/10.1111/rec.13358
Chong J, Liu P, Zhou G, Xia J (2020) Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat Protoc 15:799–821. https://doi.org/10.1038/s41596-019-0264-1
DeJong TM (1975) A comparison of three diversity indices based on their components of richness and evenness. Source. Oikos 26:222–227
Edgar R (2010) Usearch. https://www.osti.gov/servlets/purl/1137186.
Gibson KM, Nguyen BN, Neumann LM, Miller M, Buss P, Daniels S, Ahn MJ, Crandall KA, Pukazhenthi B (2019) Gut microbiome differences between wild and captive black rhinoceros – implications for rhino health. Sci Rep 9:7570. https://doi.org/10.1038/s41598-019-43875-3
Goossens B, Salgado-lynn M (2013) Advances and difficulties of molecular tools for carnivore conservation in the tropics. Raffles Bull Zool 28:43–53
Guo W, Mishra S, Wang C, Zhang H, Ning R, Kong F, Zeng B, Zhao J, Li Y (2019) Comparative study of gut microbiota in wild and captive giant pandas (Ailuropoda melanoleuca). Genes 10(10):827. https://doi.org/10.3390/genes10100827
Hannon GJ (2010) FASTX-Toolkit. http://hannonlab.cshl.edu/fastx_toolkit.
Hong J, Karaoz U, De Valpine P, Fithian W (2022) To rarefy or not to rarefy: robustness and efficiency trade-offs of rarefying microbiome data. Bioinformatics 38:2389–2396. https://doi.org/10.1093/bioinformatics/btac127
Hughes JB, Hellmann JJ (2005) The application of rarefaction techniques to molecular inventories of microbial diversity. Methods Enzymol 397:292–308
Iman MH, Kuswandi PC, Subrata SA (2024) Genetic variation of the native Rusa deer (Rusa timorensis) in Java and Bali (Indonesia) as revealed using non-invasive sampling. Biodiversitas 25(1):355–360. https://doi.org/10.13057/biodiv/d250241
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41(1):e1–e1. https://doi.org/10.1093/nar/gks808
Lin H, Das PS (2020) Analysis of microbial compositions: a review of normalization and differential abundance analysis. NPJ Biofilms Microbiomes 6(1):60. https://doi.org/10.1038/s41522-020-00160-w
Liu T, Zhao H, Wang T (2020) An empirical Bayes approach to normalization and differential abundance testing for microbiome data. BMC Bioinformatics 21:225. https://doi.org/10.1186/s12859-020-03552-z
Mahé F, Czech L, Stamatakis A, Quince C, de Vargas C, Dunthorn M, Rognes T (2021) Swarm v3: towards tera-scale amplicon clustering. Bioinformatics 38(1):267–269. https://doi.org/10.1093/bioinformatics/btab493
McKnight DT, Huerlimann R, Bower DS, Schwarzkopf L, Alford RA, Zenger KR (2019) Methods for normalizing microbiome data: an ecological perspective. Methods Ecol Evol 10:389–400. https://doi.org/10.1111/2041-210X.13115
Murray MH, Lankau EW, Kidd AD, Welch CN, Ellison T, Adams HC, Lipp EK, Hernandez SM (2020) Gut microbiome shifts with urbanization and potentially facilitates a zoonotic pathogen in a wading bird. PLoS One 15(3):e0220926. https://doi.org/10.1371/journal.pone.0220926
Pan AY (2021) Statistical analysis of microbiome data: the challenge of sparsity. Curr Opin Endocr Metab Res 19:35–40. https://doi.org/10.1016/j.coemr.2021.05.005
Paulson JN, Colin Stine O, Bravo HC, Pop M (2013) Differential abundance analysis for microbial marker-gene surveys. Nat Methods 10:1200–1202. https://doi.org/10.1038/nmeth.2658
Prabhu VR, Wasimuddin VR, Kamalakkannan R, Arjun MS, Nagarajan M (2020) Consequences of domestication on gut microbiome: a comparative study between wild Gaur and domestic Mithun. Front Microbiol 11:133. https://doi.org/10.3389/fmicb.2020.00133
Sun CH, Liu HY, Liu B, Yuan BD, Lu CH (2019) Analysis of the gut microbiome of wild and captive Père David’s deer. Front Microbiol 10:2331. https://doi.org/10.3389/fmicb.2019.02331
Sun Y, Sun Y, Shi Z, Liu Z, Zhao C, Lu T, Gao H, Zhu F, Chen R, Zhang J, Pan R, Li B, Teng L, Guo S (2020) Gut microbiota of wild and captive Alpine musk deer (Moschus chrysogaster). Front Microbiol 10:3156. https://doi.org/10.3389/fmicb.2019.03156
Sutomo, van Etten E (2021) Savanna plant communities in the wetter parts of the Indonesian archipelago. Folia Geobot 56:193–204. https://doi.org/10.1007/s12224-021-09401-y
Tang L, Li Y, Srivathsan A, Gao Y, Li K, Hu D, Zhang D (2020) Gut microbiomes of endangered Przewalski’s horse populations in short- and long-term captivity: implication for species reintroduction based on the soft-release strategy. Front Microbiol 11:363. https://doi.org/10.3389/fmicb.2020.00363
Trevelline BK, Fontaine SS, Hartup BK, Kohl KD (2019) Conservation biology needs a microbial renaissance: a call for the consideration of host-associated microbiota in wildlife management practices. Proc R Soc B 286(1895):20182448. https://doi.org/10.1098/rspb.2018.2448
Tsilimigras MCB, Fodor AA (2016) Compositional data analysis of the microbiome: fundamentals, tools, and challenges. Ann Epidemiol 26:330–335. https://doi.org/10.1016/j.annepidem.2016.03.002
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267. https://doi.org/10.1128/AEM.00062-07
Wei F, Wu Q, Hu Y, Huang G, Nie Y, Yan L (2019) Conservation metagenomics: a new branch of conservation biology. Sci China Life Sci 62:168–178. https://doi.org/10.1007/s11427-018-9423-3
Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, Lozupone C, Zaneveld JR, Vázquez-Baeza Y, Birmingham A, Hyde ER, Knight R (2017) Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5:1–18. https://doi.org/10.1186/s40168-017-0237-y
West AG, Waite DW, Deines P, Bourne DG, Digby A, McKenzie VJ, Taylor MW (2019) The microbiome in threatened species conservation. Biol Conserv 229:85–98. https://doi.org/10.1016/j.biocon.2018.11.016
Whitten T, Soeriaatmaja RE, Afiff SA (1996) The ecology of Java and Bali. Periplus Edition, Hong Kong
Willis AD (2019) Rarefaction, alpha diversity, and statistics. Front Microbiol 10:2407. https://doi.org/10.3389/fmicb.2019.02407
Xia Y, Sun J, Chen D-G (2018) Statistical analysis of microbiome data with R. Springer Singapore, Singapore
Yang H, Leng X, Du H, Luo J, Wu J, Wei Q (2020) Adjusting the prerelease gut microbial community by diet training to improve the postrelease fitness of captive-bred Acipenser dabryanus. Front Microbiol 11:488. https://doi.org/10.3389/fmicb.2020.00488
Zemanova MA (2019) Poor implementation of non-invasive sampling in wildlife genetics studies. Rethinking Ecol 4:119–132. https://doi.org/10.3897/rethinkingecology.4.32751
Zhu L, Wang J, Bahrndorff S (2021) Editorial: The wildlife gut microbiome and its implication for conservation biology. Front Microbiol 12:10–13. https://doi.org/10.3389/fmicb.2021.697499
Acknowledgements
The authors are grateful to the Directorate General of Higher Education, Research, and Technology–the Ministry of Education, Culture, Research, and Technology of the Republic of Indonesia for funding this research. The authors also thank the French Ministry of Foreign Affairs and International Development (MAEDI) and the French Ministry of Higher Education and Research (MESR) for supporting this research collaboration. The authors also acknowledge the Directorate General of Nature and Ecosystem Conservation–Ministry of Environment and Forestry of the Republic of Indonesia for issuing a sample collection permit.
Funding
Authors SAS, PY, and WTA received funding from the Directorate General of Higher Education, Research, and Technology, Indonesia, and authors MdGW, AA, and JM had support from the French Ministry of Foreign Affairs and International Development and the French Ministry of Higher Education and Research.
Author information
Authors and Affiliations
Contributions
All authors contributed to the research conception and design. Sample preparation, data collection and analysis were performed by SS and AA. The first draft of the manuscript was written by SS and AA, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
The authors affirm that no human research participants are involved in this research, and all fecal samples were collected non-invasively. Therefore, the research ethics committee of Universitas Gadjah Mada confirmed that ethical approval is not required.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Subrata, S.A., Yuda, P., Artama, W.T. et al. Rusa deer microbiota: the importance of preliminary data analysis for meaningful diversity comparisons. Int Microbiol (2024). https://doi.org/10.1007/s10123-024-00521-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10123-024-00521-x