
Abstract
The general bone anabolic effect of photobiomodulation (PBM) is largely accepted. As a result, PBM therapy is expected to be beneficial in the medical fields of dentistry and bone healing. However, most of the previous in vitro studies on PBM and bone metabolism were performed with single-cell cultures of osteoclast-lineage cells or osteoblast-lineage cells. In the present study, the bone-modulating effects of PBM were evaluated in an in vitro osteoblast/osteoclast co-culture system. Mouse bone marrow-derived macrophages (BMMs) and mouse calvarial pre-osteoblasts cells were purified and used as precursor cells for osteoclasts and osteoblasts, respectively. The PBM effects on single-cell culture of osteoclasts or osteoblasts as well as co-culture were examined by 1.2 J/cm2 low-level Ga-Al-As laser (λ = 808 ± 3 nm, 80 mW, and 80 mA; spot size, 1cm2; NDLux, Seoul, Korea) irradiation for 30 s at daily intervals throughout culture period. At the end of culture, the osteoclast differentiation and osteoblast differentiation were assessed by TRAP staining and ALP staining, respectively. The expressions of osteoclastogenic cytokines were evaluated by RT-PCR and Western blot analyses. Under the single-cell culture condition, PBM enhanced osteoblast differentiation but had minor effects on osteoclast differentiation. However, in the co-culture condition, its osteoblastogenic effect was maintained, and osteoclast differentiation was substantially reduced. Subsequent RT-PCR analyses and western blot results revealed marked reduction in receptor activator of NF-κB ligand (RANKL) expression and elevation in osteoprotegerin (OPG) expression by PBM in co-cultured cells. More importantly, these alterations in RANKL/OPG levels were not observed under the single-cell culture conditions. Our results highlight the different effects of PBM on bone cells based on culture conditions. Further, our findings suggest the indirect anti-osteoclastogenic effect of PBM, which is accompanied by a decrease in RANKL expression and an increase in OPG expression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
After the first description of low-level laser in medical practice in 1968 [1], numerous basic and clinical researchers have focused on its therapeutic benefits. Currently, photobiomodulation (PBM) is known to be capable of improving skin wounds and ulcer healing [2], enhancing angiogenesis [3], and reducing pain [4]. Despite various biological backgrounds, the encouraging positive results of PBM are considered to be mediated by “photobiostimulation,” which includes elevated RNA/DNA synthesis, increased mitochondrial respiratory activity, and altered kinase activity [5, 6]. However, its precise working mechanisms in cells and tissues remain elusive.
Bone homeostasis is maintained by balanced activities between bone-forming osteoblasts and bone-resorbing osteoclasts [7, 8]. Since the discovery of the utility of PBM as a medical therapy, significant research has been conducted to elucidate its effects on the behavior of osteoblasts and osteoclasts, which are the two important cells in bone remodeling and regeneration. In fact, Huertas et al. [9] revealed enhanced osteoblast proliferation upon PBM in the human osteosarcoma cell line, MG63. More specifically, PBM increased the secretion of growth factors, including insulin-like growth factor I (IGF-I), basic fibroblast growth factor (bFGF), and receptor of IGF-I (IGFBP3), from osteoblasts [10]. Therefore, it is assumed that one of the underlying mechanisms for the PBM-induced positive regulation of osteoblast differentiation is mediated by the autocrine action of growth factors. However, Bouvet-Gerbettaz et al. revealed that PBM had minor or no effects on bone cell behavior [11]. Furthermore, PBM was not found to alter osteoblast or osteoclast differentiation. Although such controversy may arise from differences in the experimental context, the overall bone healing effects of PBM have been continually proven and accepted by researchers [12, 13].
Under in vitro and in vivo conditions, osteoclasts and osteoblasts communicate with each other, and this coupling mechanism contributes to the fine regulation of bone remodeling [14, 15]. Through the reciprocal coupling between osteoclasts and osteoblasts, the bone-forming activity of osteoblasts meets the demands induced by osteoclast-mediated bone resorption, which allows the replacement of lost bone in the remodeling process [15]. Osteoclast–osteoblast communication occurs in at least three modes: direct cell-to-cell contact, soluble paracrine factor secretion (clastokine), and osteoclastic bone resorption-mediated liberation of bone matrix-stored growth factors [14]. As a result, it is reasonable to assess the anabolic or catabolic effects of particular drugs or therapeutic methods by considering osteoclast–osteoblast coupling. However, most prior studies on the effects of PBM on bone homeostasis were conducted with either osteoclast or osteoblast lineage cells alone.
Therefore, in the present study, we evaluated the bone anabolic effects of PBM using primary mouse pre-osteoclasts and primary mouse calvarial pre-osteoblasts via a single culture of each cell or co-culture system.
Materials and methods
Osteoclast differentiation
Bone marrow-derived macrophages (BMMs) were isolated and used as precursor cells for osteoclastogenesis. The isolation methods and cultures of BMMs were as described previously [16, 17]. In brief, the femora and tibiae were extracted from 5-week-old female ICR mice, and whole bone marrow cells were flushed out from the marrow spaces of long bones with Hank’s balanced salt solution (HBSS; WelGENE Inc., Daegu, Korea). The flushed whole bone marrow cells were cultured on 100-mm cell culture grade dishes in complete α-modified Eagle’s medium (α-MEM; WelGENE Inc., Daegu, Korea) for 12 h. During the 12-h culture, unnecessary fibroblasts and stromal cells became adherent. The floating cells were collected and further cultured on petri dishes in the presence of macrophage colony-stimulating factor (M-CSF, 30 ng/mL; PeproTech, Rocky Hill, NJ, USA) for 3 days. At the end of the culture, adherent BMMs were detached by scraping and were used as osteoclast precursor cells. Osteoclast differentiation was achieved by culturing BMMs (4 × 104 cells per well in a 48-well plate) in an osteoclastogenic medium (30 ng/mL M-CSF + 100 ng/mL RANKL) for 4 days. RANKL was purchased from PeproTech (Rocky Hill, NJ, USA). After culture, multinucleated osteoclasts were stained for tartrate-resistant acid phosphatase activity (TRAP activity) using a leukocyte acid phosphatase kit (Sigma-Aldrich, St Louis, MO, USA), according to the manufacturer’s instructions. Subsequently, the number of TRAP-positive multinucleated cells (TRAP + MNCs) with ≥ 3 nuclei was counted as osteoclasts under the light microscope (× 100 magnification with Olympus-BX51 microscope [Olympus, Center Valley, PA]). All animal experiments were approved by the Committee on the Care and Use of Animals in Research at Pusan National University (PNU-2018–2037).
Osteoblast differentiation
Primary pre-osteoblasts (calvarial osteoblasts, Cal-OBs) were prepared from 1-day-old mice and used as precursor cells for osteoblast differentiation [18, 19]. The dissected calvarias were sequentially enzyme-digested with 0.1% collagenase and 0.2% dispase for 15 min at 37 °C under gentle shaking conditions. The prepared calvarial osteoblasts were allowed to differentiate into mature osteoblasts by culturing cells in osteogenic medium (OM, α-MEM supplemented with 10 mM β-glycerophosphate and 100 μ g/mL ascorbic acid) for 7 days. Osteogenic differentiation was confirmed by alkaline phosphatase (ALP) staining (leukocyte alkaline phosphatase kit; Sigma-Aldrich, St. Louis, MO, USA). The enzyme activity of ALP was quantitatively measured using the ALP assay kit from TAKARA BIO, Inc. (Shiga, Japan).
Co-culture of osteoclast–osteoblast
In some experiments, the biological effect of PBM was examined under osteoclast–osteoblast co-culture conditions. First, prepared calvarial osteoblasts were seeded at 2 × 104 cells per well in a 48-well plate. On the following day, BMMs were seeded on the plated calvarial osteoblasts (2 × 105 cells per well) in the presence of 10 nM 1α,25-dihydroxyvitaminD3 (VitD3) and 1 μM prostaglandinE2 (PGE2). Although the exact period required for the complete differentiation of osteoclasts or osteoblasts varied between experiments, mature osteoclasts or osteoblasts were generally observed after 5–7 days of co-culture. At the end of culture, the extent of osteoclastogenesis or osteoblastogenesis was evaluated by TRAP staining or ALP staining, respectively.
Photobiomodulation (PBM)
Photobiomodulation experiments were performed as described previously [20, 21]. In brief, laser irradiation was delivered with a low-level gallium-aluminum-arsenide laser (Ga-Al-As laser; λ = 808 ± 3 nm, 80 mW; 80 mA; spot size, 1 cm2; NDLux, Seoul, Korea). Laser irradiation was performed in continuous mode on an aseptic clean bench (under dark conditions), and the laser handpiece was fixed in vertical direction to each well (5 cm above from the bottom of the culture plate). The cell culture plate was moved sequentially to irradiate one well at a time. The laser irradiation was applied for 30 s at daily intervals throughout the culture period, and the delivered energy was 1.2 J/cm2 per irradiation. The energy doses that were received by cells were measured at cell surface level using a laser power meter (Coherent Inc., San Jose, CA, USA). PBM was initiated on the day after cell seeding, and the same irradiation protocols were used for both single-cell culture and co-culture experiments. During the irradiation time, the control plates were placed under room light for the same duration as the PBM-treated plates.
Conditioned media (CM) collection from the co-culture
Calvarial pre-osteoblasts and BMMs were co-cultured in the co-culture medium (complete α-MEM containing 10 nM VitD3 and 1 μM PGE2) for 6 days and PBM treated with a low-level laser each day. On day 6, the culture medium was replaced with fresh α-MEM and incubated for 24 h. Thereafter, the CM was collected and immediately centrifuged at 1500 rpm for 5 min to discard superfluous cell remnants. The prepared CM is used for functional osteoclastogenic activity assay as shown in Fig. 4.
Reverse transcriptase polymerase-chain reaction (RT-PCR) and quantitative real-time PCR analyses
Qualitative RT-PCR analyses and quantitative real-time PCR analyses were performed following a standard procedure. Briefly, for RT-PCR, total RNA was purified from co-cultured cells (at co-culture day 4) using TRIzol Reagent (Invitrogen, Waltham, MA, USA). Thereafter, 1.5 μg of RNA was reverse transcribed into cDNA with Superscript II (Invitrogen), according to the manufacturer’s instructions. The mRNA expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. The numbers of PCR amplification cycles were determined to be in a linear range of amplification. Eighteen cycles were run for GAPDH, while 25–30 cycles were carried out for the remaining RNAs using T100 Thermal Cycler (Bio-Rad, Hercules, CA). The PCR products were run on a 2% agarose gel and observed under a UV illuminator, UView™ Mini Trans-illuminator (Bio-Rad, Hercules, CA). The sequences of the RT-PCR primers were: TGF-β1, 5′-GCTTCAGACAGAAACTCACT-3′ (forward) and 5′-GAACACTACTACATGCCATTA T-3′ (reverse); IL-6, 5′-TGTGCAATGGCAATTCTGAT-3′ (forward) and 5′-TCCTTCCTACCCCAATTTCC-3′ (reverse); IL-1α, 5′-GCAAGCTATGGCTCACTTCA-3′ (forward) and 5′-CGTCAGGCAGAAGTTTGTCA-3′ (reverse); M-CSF, 5′-GCTTGGCTTGGGATGATTCT-3′ (forward) and 5′-GGCTTTTGGTCCTCTGTTGA-3′ (reverse); RANKL, 5′-CCAGCATCAAAATCCCAAGT-3′ (forward) and 5′-CCCCTTCAGATGATCCTTC-3′ (reverse); OPG, 5′-TGCAGTACGTCAAGCAGGAG-3′ (forward) and 5′-TGACCTCTGTGAAAACAGC-3′ (reverse); GAPDH, 5′-GGAGAGTGTTTCCTCGTCCC-3′ (forward) and 5′-AGACACCAGTAGACTCCACG-3′ (reverse). For quantitative real-time PCR, reversed transcribed 1 μg of cDNAs were analyzed with KAPA SYBR FAST qPCR kit (Kapa Biosystems, Wilmington, MA, USA) using ABI 7500 instruments (Applied Biosystems, Forster City, CA, USA) by following PCR conditions: 40 cycles of 3 s denaturation at 95 ℃ and 30 s amplifications at 60 ℃. The mRNA expression levels of RANKL and OPG were normalized to that of hypoxanthine phosphoribosyl transferase (HPRT). The primer sets used in real-time PCR analyses were as follows: RANKL, 5′-AGGCTGGGCCAAGATCTCTA-3′ (forward) and 5′-GTCTGTAGGTACGCTTCCCG-3′ (reverse); OPG, 5′-TCCTGGTGCTCCTGGACAT-3′ (forward) and 5′-AGGGCAAGGGACACACAATG-3′ (reverse); HPRT, 5′-CCTAAGATGATCGCAAGTTG-3′ (forward) and 5′-CCACAGGGACTAGAACACCTGCTAA-3′ (reverse). In this study, all of the primer sets were designed and validated using web-based Primer-BLAST program (https://www.ncbi.nlm.nih.gov/tools/primer-blast), and the quantitative results were obtained from three independent experiments performed in triplicate.
Western blotting
Western blotting was performed according to a standard procedure. Briefly, co-cultured cells (at co-culture day 4) were lysed with RIPA lysis buffer (50 mM Tris, pH 8.0; 150 mM NaCl; 0.5% sodium deoxycholate; 1.5 mM MgCl2; 1% Triton X-100; and protease inhibitor cocktail). Thereafter, the protein concentration in the lysates was determined with a detergent-compatible protein assay kit (Bio-Rad Laboratories, CA, USA), and 30–40 μg of cell lysate was resolved via electrophoresis on 8–10% SDS–polyacrylamide gel (SDS-PAGE). The resolved protein bands were transferred onto nitrocellulose membranes, blocked with 5% skim milk for 1 h, and probed with primary antibodies against M-CSF (Abcam, Cambridge, MA, USA), RANKL (Santa Cruz Biotechnology, Santa Cruz, CA, USA), OPG (Santa Cruz Biotechnology, Santa Cruz, CA, USA), or β-actin (Sigma-Aldrich, St Louis, MO, USA). The immunoreactivity of the membranes was detected with chemiluminescence reagents. The western blotting results were obtained from three independent experiments. Among the multiple results, only the representative results were shown, and the band images were quantified with ImageJ program (ver 1.50i, National Institutes of Health, Bethesda, MD, USA) by comparing the band intensities (relative band intensities normalized to that of control actin blot).
Statistics
Data were obtained from at least three independent experiments performed in triplicate. Student’s t test was used to determine the significance of differences between two groups. A p value < 0.05 was regarded as statistically significant and is denoted with an asterisk in the text.
Results
PBM leads to augmented osteoblast differentiation but has minor effects on osteoclast differentiation in a single-cell culture system
First, we sought to determine the effects of PBM on the differentiation of osteoblasts or osteoclasts using an independent single-cell culture system. After purifying pre-osteoblasts (Cal-OBs) from the calvariae of newborn mice and mouse BMMs from the long bones of 5-week-old mice, we employed these cells as precursor cells for osteoblasts and osteoclasts, respectively. As shown in Fig. 1a, Cal-OBs are differentiated into ALP-positive osteoblasts after culturing in osteogenic medium (OM). PBM remarkably enhanced the formation of ALP-positive osteoblasts compared to the treatment with OM alone. However, osteoclastogenesis was not affected by the presence of PBM (Fig. 1b). Therefore, we concluded that PBM could promote osteoblast differentiation but will not affect osteoclast differentiation, at least in the context of a single-cell culture.

Photobiomodulation (PBM) significantly increased osteoblast differentiation but had a minor effect on osteoclast differentiation under the single-cell culture condition. a Calvarial pre-osteoblast cells purified from newborn mice were cultured in an osteogenic medium (OM; α-MEM including 100 μg/mL ascorbic acid plus 10 mM β-glycerophosphate) and daily treated with PBM (daily irradiation of low-level laser). After 5 days of culture, the extent of osteoblast differentiation was visualized via ALP staining, and ALP enzyme activities were quantitatively examined (*p < 0.05 versus OM only). b Mouse bone marrow-derived macrophages (BMMs) were differentiated into osteoclasts in the osteoclastogenic medium (α-MEM containing 30 ng/mL M-CSF + 100 ng/mL RANKL) in the presence or absence of daily PBM for 4 days. Mature osteoclasts were stained to evaluate TRAP activity, and the number of TRAP-positive multinucleated cells (TRAP + MNCs) was counted. All quantitative data are presented as mean ± standard deviation (SD) (*p < 0.05 versus M-CSF + RANKL)
PBM attenuates osteoclast differentiation in the co-culture system
Although the single-cell culture system is one of the generally recognized first steps in determining the clinical advantages of newly developed medical devices, the actual tissue of an organism is composed of different cells. In recent studies, a close connection has been well established between the activities of osteoblasts and osteoclasts [14, 15]. Thus, we proceeded to determine the effect of PBM on osteoblast/osteoclast differentiation in a co-culture system. In this experiment, BMMs were seeded on Cal-OBs in the presence of anabolic stimulus (prostaglandin E2 and vitamin D3) with PBM for 7 days. Thereafter, differentiation was subsequently assessed using ALP or TRAP staining (Fig. 2a). Similar to the result of the single-cell culture (Fig. 1a), osteoblast differentiation was increased upon PBM treatment; however, osteoclast differentiation was substantially decreased in the presence of PBM (Fig. 2b). Such findings suggest that there might be alterations in the responsiveness of cells to the same PBM based on the cell culture context.

PBM reduced osteoclast formation under the co-culture condition. a BMMs were co-cultured with calvarial pre-osteoblasts in the co-culture medium containing 10 nM of VitD3 and 1 μM of prostaglandinE2. During the 7 days of the co-culture period, PBM was applied daily. At the end of the culture, osteoblast differentiation and osteoclast differentiation were assessed via ALP staining and TRAP staining, respectively. b Cells were cultured as described in (a), and the osteoblastic ALP enzyme activities and TRAP-positive multinucleated osteoclasts were quantified (*p < 0.05 versus PGE2 + VitD3 group)
PBM reduces RANKL/OPG ratio in the co-culture condition
To further shed light on the underlying mechanisms of the PBM-mediated decrease in osteoclastogenesis during co-culture, we sought to determine the effect of PBM on the expression of osteoclastogenic cytokines in co-culture conditions. The mRNA expression levels of a series of osteoclastogenic cytokines, which are known to support osteoclast differentiation [22, 23], were determined by RT-PCR. Based on our findings, the mRNA expression levels of transforming growth factor-β (TGF-β), interleukin-6 (IL-6), interleukin-1 (IL-1), and macrophage colony-stimulating factor (M-CSF) were comparable in the presence or absence of PBM (Fig. 3a). However, the expression level of RANKL was markedly reduced upon PBM compared to treatment with PGE2 plus VitD3 (Fig. 3a and b). In contrast, the expression level of OPG was upregulated in the PBM-treated group (Fig. 3a and b). Notably, the quantitative real-time PCR results show a significant decrease in RANKL/OPG ratio upon PBM treatment. The RANKL/OPG ratio was 3.4 ± 0.75 at PGE2 plus VitD3-treated group, but that was reduced to 0.58 ± 0.03 upon PBM treatment (Fig. 3b). These altered expression levels of RANKL and OPG were also confirmed by western blot analyses (Fig. 3c–e). Aligning with the mRNA expression results, the increase in RANKL and decrease in OPG were prominent at the protein level in the PBM-treated group. As RANKL is known to be essential for osteoclast differentiation and OPG is well appreciated as a decoy receptor for RANKL [23, 24], the PBM suppression of osteoclastogenesis in the co-culture might be due to the decreased RANKL/OPG ratio in the co-culture context.

PBM substantially decreased RANKL expression and increased OPG expression under the co-culture condition. a BMMs and calvarial osteoblasts were co-cultured for 4 days in the presence or absence of PBM. The mRNA expression levels of different cytokines (TGF-β, IL-6, IL-1, M-CSF, RANKL, and OPG) were determined by RT-PCR. GAPDH served as a loading control. b Cells were cultured and treated as in (a) and the mRNA expression levels of RANKL as well as OPG were determined by quantitative real-time PCR. The gene expression levels were normalized to that of HPRT. c Cells were co-cultured as described in (a), and whole-cell lysates were prepared. The protein expression levels of M-CSF, RANKL, and OPG were examined by western blotting. Actin served as a loading control. d, e The relative band intensities of western blotting performed in triplicates as in (c) were normalized to that of the control band (actin) and are presented as statistical graphs (*p < 0.05 versus PGE2 + VitD3 group)
The CM from the PBM-treated co-culture exhibits functionally-reduced osteoclastogenic activity
To functionally verify the reduced RANKL/OPG ratio during PBM treatment in the co-culture, we harvested the CM from PBM-treated co-cultured cells and determined the osteoclastogenic activity of CM with independent osteoclastogenesis culture. As shown in Fig. 4a, co-cultures of BMMs plus Cal-OB are performed, and PBM is administered daily for 6 days. The soluble factors secreted from the co-cultured cells were collected in empty α-MEM medium for an additional day. The collected CM was administered to separate pre-osteoclast cultures, and its pro-osteoclastogenic activity was assessed. In accordance with the results shown in Fig. 3 (i.e., reduced RANKL/OPG ratio owing to PBM treatment), the CM obtained from the PBM-treated co-culture had reduced pro-osteoclastogenic activity compared to the CM obtained from the non-PBM control (Fig. 4b). These data further support the notion that PBM could exert an anti-osteoclastogenic effect by indirectly lowering critical soluble factors for osteoclast differentiation.

The osteoclastogenic soluble cytokine expression levels were functionally reduced following PBM treatment. a Schematic diagram of the experiments. BMMs and calvarial osteoblasts were co-cultured in control medium (complete α-MEM) or co-culture medium (complete α-MEM containing 10 nM VitD3 + 1 μM prostaglandinE2), with daily treatment of PBM for 6 days. At day 6, the medium was replaced with fresh α-MEM, and the conditioned medium (CM) was harvested after 24 h. The osteoclastogenic activities of the CM from the co-culture condition were evaluated with an independent osteoclast differentiation system. BMMs were primed to pre-osteoclasts (pre-OCs) via a 2-day culture with M-CSF and RANKL. At day 2, the culture medium was replaced with CM, and pre-OCs were allowed to differentiate into mature osteoclasts for 2 additional days. b Cells were cultured as described in (a) and representative TRAP-stained images are presented. The number of TRAP-positive osteoclasts was counted (*p < 0.05 versus PGE2 + VitD3 group)
Discussion
Following the invention of the laser by Theodor Maiman, its usefulness has been assessed in several fields, including industry as well as medicine. Although there are several approaches to classifying lasers, their use is generally classified by the amount of transmitting energy they emit (i.e., high-, medium-, and low-energy lasers) [6, 25]. High- and medium-energy lasers are mainly applied in the surgery and oncology specialties, respectively [6]. The relatively safe and non-destructive nature of low-level lasers has promoted their extensive application in medical practice. Different beneficial effects of PBM via low-level laser, such as skin wound healing, pain relief, accelerated angiogenesis, and bone healing, have been reported [6, 26]. However, despite these encouraging results, the exact molecular action of PBM on cells and tissues remains elusive.
The effectiveness of PBM has been extensively evaluated in different studies performed in the field of bone biology. Huertas et al. [9] observed increased proliferation of osteoblasts via PBM using the human osteosarcoma cell line, MG-63. Furthermore, Saygun et al. [10] examined the elevated expression of growth factors (bFGF, IGF-I, IGFBP3) using a PBM during osteogenic differentiation of human mesenchymal stem cells (hMSCs). Human dental pulp stem cells and adipose-derived stem cells, which have the potency to differentiate into the osteogenic lineage, showed significantly enhanced proliferation rate upon PBM relative to the non-PBM-treated cells [27, 28]. Similarly, the effects of PBM on osteoclasts, which are specific bone-resorbing cells, have also been described. Aihara et al. reported the osteoclastogenesis-stimulating effect of PBM using rat osteoclast precursor cells [29]. In this study, PBM facilitated the expression of RANK, the receptor for RANKL, which resulted in the upregulation of osteoclast differentiation in vitro. However, other researchers have shown that murine osteoclast differentiation is not affected by PBM in a comparable in vitro condition [11]. In the present study, PBM was not found to influence the differentiation of osteoclasts from mouse BMMs in the single-cell culture condition (Fig. 1b). The differences between the results might be due to the diversity of PBM and cell culture condition, specifically the dissimilarity in the power density of the laser, origin of experimental cells, and the type of cell cultures.
Bone homeostasis is finely regulated by the strict coupling of osteoclast-mediated bone resorption and osteoblast-mediated bone formation. The osteoclast and osteoblast lineage cells continually interact with each other via cell-to-cell contact, soluble paracrine factors, and growth factors liberated from the bone matrix [15]. The essential osteoclastogenic cytokine, RANKL (also called TRANCE), is produced by osteoblast lineage cells. However, osteoclast precursor cells express RANK, the receptor for RANKL [30]. The ligation of RANKL-RANK initiates the osteoclastogenic signaling cascades that converge on the expression of pro-osteoclastogenic transcription factors, such as NF-kB, AP-1, and NFATc1 [30, 31]. Osteoblasts also express a soluble decoy receptor for RANKL, OPG, which antagonizes RANKL signaling by masking RANKL [30, 31]. Therefore, the RANKL/OPG ratio is currently believed to be the most important rate-limiting factor for bone quality and integrity. In our experiments, a prominent decrease in the RANKL/OPG ratio was observed in PBM-treated co-cultured cells (Fig. 3b). Because our co-culture system did not include the extrinsic administration of RANKL or OPG, osteoclast differentiation was entirely dependent on the RANKL produced by osteoblasts. Accordingly, the decreased osteoclast formation reflects the decreased production of RANKL from osteoblasts in the PBM group (Fig. 2a). Such findings suggest that PBM might exert an indirect anti-osteoclastogenic effect by reducing the expression of RANKL produced from osteoblasts. More notably, this phenomenon is contingent on the cell culture context. Consistent with our results, Xu et al. reported a decreased mRNA expression of RANKL by PBM (low-intensity pulsed laser irradiation) in rat calvarial cells [32]. However, Incerti et al. observed opposing results; they revealed a trend of rapid and transient increase in RANKL/OPG ratio following PBM in the human osteoblast-like cells, Saos-2 [33]. As aforementioned, this controversy might arise from differences in the experimental contexts, specifically differences in the PBM parameters and cells.
The mechanism of action employed by PBM to reduce RANKL expression in osteoblasts remains to be clarified. In fact, despite the existence of numerous studies on PBM therapy, its precise therapeutic mechanism remains unclear. However, its effects are generally described according to “photobiostimulation,” which encompasses changes in reactive oxygen species (ROS) or adenosine triphosphate (ATP), modifications of several kinase activities, and alterations in ion-channel activities [2, 6]. Because of these extensive effects, we have opted to focus on previous studies that showed that PBM triggered an increase in ROS production [34, 35]. ROS signaling is known to induce RANKL expression in mouse osteoblasts and human MG63 cells [36, 37]. In accordance with these results, PBM was found to enhance RANKL expression under the co-culture condition (Fig. 3). However, the PBM-mediated upregulation of ROS, which is crucial for RANKL expression, must be clarified via future studies.
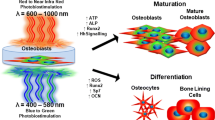
In the present study, we revealed the indirect inhibition of osteoclast differentiation using PBM (Fig. 5). Although PBM had minor effects on osteoclast differentiation in the single-cell culture of osteoclast precursor cells, it reduced RANKL expression in osteoblasts in a co-culture condition, which led to a decrease in osteoclastogenesis. Such finding signifies the need to comprehensively understand bone cell behavior based on precise cell–cell interaction; this could provide a prime target for the development of bone-anabolic medical devices.

Illustration of the study’s results. Under the single-cell culture condition, PBM enhanced osteoblast differentiation but had a minor effect on osteoclast differentiation. However, in the co-culture condition that more reflects the bio-mimetic environment, PBM decreased the expression of the pro-osteoclastogenic cytokine, RANKL, and increased that of the anti-osteoclastogenic cytokine, OPG, in osteoblasts, resulting in an indirect decrease in osteoclast differentiation
Conclusion
In summary, the photobiomodulation enhanced osteoblastogenesis but had little effects on osteoclastogenesis under the single-cell culture of each cell. However, in the co-culture condition, the osteoclastogenesis was markedly reduced by the presence of photobiomodulation through the downregulation of pro-osteoclastogenic RANKL expression and upregulation of anti-osteoclastogenic OPG expression. Our finding reveals an indirect anti-osteoclastogenic effect of photobiomodulation as well as emphasizes the importance of cell-to-cell communications in photobiomodulation study.
Data availability
The data presented in this study are available within this article.
Code availability
Not applicable.
References
Mester E, Szende B, Gartner P (1968) The effect of laser beams on the growth of hair in mice. Radiobiol Radiother 9:621–626
Woodruff L, Bounkeo J, Brannon W, Dawes K, Barham C, Waddell D, Enwemeka C (2004) The efficacy of laser therapy in wound repair: a meta-analysis of the literature. Photomed Laser Surg 22:241–247
Cury V, Moretti A, Assis L, Bossini P, de Souza CJ, Neto C, Fangel R, de Souza H, Hamblin M, Parizotto N (2013) Low level laser therapy increases angiogenesis in a model of ischemic skin flap in rats mediated by VEGF, HIF-1α and MMP-2. J Photochem Photobiol B 125:164–170
Bjordal J, Johnson M, Iversen V, Aimbire F, Lopes-Martins R (2006) Low-level laser therapy in acute pain: a systematic review of possible mechanisms of action and clinical effects in randomized placebo-controlled trials. Photomed Laser Surg 24:158–168
AlGhamdi K, Kumar A, Moussa N (2012) Low-level laser therapy: a useful technique for enhancing the proliferation of various cultured cells. Lasers Med Sci 27:237–249
Rola P, Doroszko A, Derkacz A (2014) The use of low-level energy laser radiation in and clinical research. Adv Clin Exp Med 5:835–842
Boyce BF (2013) Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res 10:860–867
Harada S, Rodan GA (2003) Control of osteoblast function and regulation of bone mass. Nature 6937:349–355
Huertas R, Luna-Bertos E, Ramos-Torrecillas J, Leyva F, Ruiz C, Garcia-Martinez O (2013) Effect and clinical implications of the low-energy diode laser on bone cell proliferation. Biol Res Nurs 2:191–196
Saygun I, Nizam N, Ural A, Serdar M, Avcu F, Tozum T (2012) Low-level laser irradiation affects the release of basic fibroblast growth factor (bFGF), insulin-like growth factor-I (IGF-I), and receptor of IGF-I (IGFBP3) from osteoblasts. Photomed Laser Surg 30:149–154
Bouvet-Gerbettaz S, Merigo E, Rocca JP, Carle GF, Rochet N (2009) Effects of low-level laser therapy on proliferation and differentiation of murine bone marrow cells into osteoblasts and osteoclasts. Lasers Surg Med 4:291–297
Nissan J, Assif D, Gross M, Yaffe A, Binderman I (2006) Effect of low intensity laser irradiation on surgically created bony defects in rats. J Oral Rehabil 33:619–624
Khadra M, Ronold H, Lyngstadaas S, Ellingsen J, Haanaes H (2004) Low-level laser therapy stimulates bone-implant interaction: an experimental study in rabbits. Clin Oral Implants Res 15:325–332
Matsuo K, Irie N (2008) Osteoclast-osteoblast communication. Arch Biochem Biophys 2:201–209
Martin TJ, Sims NA (2005) Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med 2:76–81
Kim HJ, Lee Y, Chang EJ, Kim HM, Hong SP, Lee ZH, Ryu J, Kim HH (2007) Suppression of osteoclastogenesis by N, N-dimethyl-D-erythro-sphingosine: a sphingosine kinase inhibition-independent action. Mol Pharmacol 2:418–428
Kim HJ, Prasad V, Hyung SW, Lee ZH, Lee SW, Bhargava A, Pearce D, Lee Y, Kim HH (2012) Plasma membrane calcium ATPase regulates bone mass by fine-tuning osteoclast differentiation and survival. J Cell Biol 7:1145–1158
Ryu J, Kim HJ, Chang EJ, Huang H, Banno Y, Kim HH (2006) Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J 24:5840–5851
Lee Y, Ha J, Kim HJ, Kim YS, Chang EJ, Song WJ, Kim HH (2009) Negative feedback Inhibition of NFATc1 by DYRK1A regulates bone homeostasis. J Biol Chem 48:33343–33351
Lee JY, Kim IR, Park BS, Kim YD, Chung IK, Song JM, Shin SH (2015) Effect of low-level laser therapy on oral keratinocytes exposed to bisphosphonate. Laser Med Sci 30:635–643
Shin SH, Kim KH, Choi NR, Kim IR, Park BS, Kim YD, Kim UK, Kim CH (2016) Effect of low-level laser therapy on bisphosphonate-treated osteoblasts. Maxillofac Plast Reconstr Surg 38:48
Zupan J, Jeras M, Marc J (2013) Osteoimmunology and the influence of pro-inflammatory cytokines on osteoclasts. Biochem Med 1:43–63
Amarasekara DS, Yun H, Kim S, Lee N, Kim H, Rho J (2018) Regulation of osteoclast differentiation by cytokine networks. Immune Netw 1:e8
Martin TJ, Sims NA (2015) RANKL/OPG; Critical role in bone physiology. Rev Endocr Metab Disord 2:131–139
de Freitas LF, Hamblin MR (2016) Proposed mechanisms of photobiomodulation or low-level light therapy. IEEE J Sel Top Quantum Electron 3
Bayat M, Virdi RF, Chien S (2018) Comparison of the in vitro effects of low-level laser therapy and low-intensity pulsed ultrasound therapy on bony cells and stem cells. Prog Biophys Mol Biol 133:36–48
Eduardo Fde P, Bueno DF, de Freitas PM, Marques MM, Passos-Bueno MR, Eduardo Cde P, Zatz M (2008) Stem cell proliferation under low intensity laser irradiation: a preliminary study. Lasers Surg Med 6:433–438
Mvula B, Mathope T, Moore T, Abrahamse H (2008) The effect of low level laser irradiation on adult human adipose derived stem cells. Lasers Med Sci 3:277–282
Aihara N, Yamaguchi M, Kasai K (2006) Low-energy irradiation stimulates formation of osteoclast-like cells via RANK expression in vitro. Lasers Med Sci 1:24–33
Hamdy NA (2007) Targeting the RANK/RANKL/OPG signaling pathway: a novel approach in the management of osteoporosis. Curr Opin Investig Drugs 4:299–303
Asagiri M, Takayanagi H (2007) The molecular understanding of osteoclast differentiation. Bone 2:251–264
Xu M, Deng T, Mo F, Deng B, Lam W, Deng P, Zhang X, Liu S (2009) Low-intensity pulsed laser irradiation affects RANKL and OPG mRNA expression in rat calvarial cells. Photomed Laser Surg 2:309–315
Incerti Parenti S, Checchi L, Fini M, Tschon M (2014) Different doses of low-level laser irradiation modulate the in vitro response of osteoblast-like cells. J Biomed Opt 10:108002
Karu TI, Pyatibrat LV, Kalendo GS (2004) Photobiological modulation of cell attachment via cytochrome c oxidase. Photochem Photobiol Sci 2:211–216
Lavi R, Shainberg A, Friedmann H, Shneyvays V, Rickover O, Eichler M, Kaplan D, Lubart R (2003) Low energy visible light induces reactive oxygen species generation and stimulates an increase of intracellular calcium concentration in cardiac cells. J Biol Chem 42:40917–40922
Bai XC, Lu D, Liu AL, Zhang ZM, Li XM, Zou ZP, Zeng WS, Cheng BL, Luo SQ (2005) Reactive oxygen species stimulates receptor activator of NF-kappaB ligand expression in osteoblast. J Biol Chem 17:17497–17506
Zhang Y, Zhang Y, Kou J, Wang C, Wang K (2014) Role of reactive oxygen species in angiotensin II: induced receptor activator of nuclear factor-κB ligand expression in mouse osteoblastic cells. Mol Cell Biochem 1–2:249–255
Funding
This study was supported by the Dental Research Institute (PNUDH DRI-2019–02), Pusan National University Dental Hospital.
Author information
Authors and Affiliations
Contributions
Conceptualization and supervision, Hyung Joon Kim and Jae-Yeol Lee; methodology and validation, Ji-Un Hong and Jin-Ju Kwon; data curation, Soon Chul Heo and Sang-Hun Shin; writing-original draft preparation, Hyung Joon Kim and Ji-Un Hong. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
This study was conducted under the guidelines of the Declaration of Helsinki, and approved by the Pusan National University Institutional Animal Care and Use Committee (PNU-IACUC, approved on March 2, 2020).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
All authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hong, JU., Kwon, JJ., Heo, S.C. et al. Effects of photobiomodulation on bone remodeling in an osteoblast–osteoclast co-culture system. Lasers Med Sci 37, 1049–1059 (2022). https://doi.org/10.1007/s10103-021-03352-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-021-03352-8