
Abstract
Low-level laser therapy (LLLT) is used in the treatment of chemoradiotherapy- or radiotherapy-induced oropharyngeal mucositis (ORM). In head and neck cancer, tumor cells may lie in the LLLT irradiation field, and LLLT might promote tumor progression. We therefore investigated the effect of LLLT on proliferation, cell cycle distribution, and apoptosis in a human oral carcinoma cell line (SCC-25), non-malignant epithelial cells (BEAS-2B), and fibroblasts in vitro. The cell lines were subjected to LLLT on three consecutive days for 15 min. Cell proliferation was assessed using the MTT assay, cell cycle distribution by flow cytometry and propidium-iodide DNA staining, and apoptosis using an Annexin V-FITC assay. Controls were sham-treated, but not exposed to the laser treatment. LLLT treatment resulted in increased fibroblast proliferation (p < 0.001), whereas decreased cell proliferation was observed after LLLT treatment of BEAS-2B (p = 0.003) and SCC-25 cells (p < 0.001). In SCC-25 cells, an increased percentage of S-phase cells and decreased percentage of G1-phase cells were observed (p < 0.001). Moreover, a proapoptotic effect of LLLT was observed in SCC-25 cells (p = 0.02). LLLT did not exhibit a tumor-promoting effect in this in vitro study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oropharyngeal mucositis (ORM) is a common and severe early complication in the treatment of head and neck cancer patients undergoing chemoradiotherapy or radiotherapy alone [1]. ORM may occur as early as 1 week after the onset of therapy and may lead to pain, dysphagia, dehydration, micronutrient deficiencies, weight loss, and potentially life-threatening aspiration. Delay of anticancer treatment, a common consequence of ORM, affects treatment outcome and disease prognosis [2]. Oral hygiene, as well as anti-inflammatory and analgesic therapies, is currently the most established strategies in prevention and treatment of ORM [3]. In the pathogenesis of ORM, radiation and chemotherapy trigger cellular and DNA damage in the epithelium as well as in the submucosa. Irradiation-induced reactive oxygen species initiate a complex downstream cascade including nuclear factor-κB (NF-κB) activation and release of proinflammatory cytokines including TNF-α, IL-1β, and IL-6. Positive-feedback loops lead to signal amplification resulting in tissue injury via matrix metalloproteinases (MMPs) [4]. It is, however, still unclear as to what extent airway epithelial cells contribute to the production of inflammatory cytokines. Interestingly, a recent study revealed no inflammatory response of airway epithelia following irradiation with 2, 5, and 8 Gy [5].
Low-level laser therapy (LLLT) is the application of monochromatic coherent light at energy levels low enough that temperature elevation in the tissue is below 0.1°C. Continuously emitting low-power laser sources in the range of 1 to 500 mW are usually employed. The wavelength is typically in the red or near infrared spectrum (600–1,000 nm). Helium–neon lasers or diode lasers (e.g., gallium–aluminum–arsenide) are frequently used. In ORM, the energy densities (doses) applied to the mucosa range between 1.8 and 10 J/cm2. LLLT is administered once or several times on consecutive days or each second or third day. Usually, 9–12 spots are irradiated at different sites of the oral mucosa in contact mode or with a distance of approximately 1 cm between the laser probe and the mucosal surface. Recently, Huang et al. reported that not only the irradiation dose (J/cm2), but also the dose rate, influences the biological and clinical effects. The dose rate depends on the power density (irradiance, W/cm2) and exposure time. Keeping the total irradiation dose constant, the authors observed differential effects with high-power densities and short exposure times compared to low-power densities and long exposure times [6]. Although the total dose was similar to that used in various clinical trials on LLLT in ORM, the dose rates differed substantially.
In animal studies, LLLT was shown to promote tissue repair and exert anti-inflammatory effects in ORM [7, 8]. LLLT was also reported to be effective in the prevention of ORM. In clinical studies, higher grade mucositis occurred less frequently and pain scores were significantly reduced [9–11]. Molecular mechanisms of LLLT were found to involve mitochondrial pathways [6]. In vitro, cell proliferation has been a frequently observed effect after LLLT in various cell types including stem, mesenchymal, muscle, and blood cells [12]. Furthermore, LLLT might also affect specific proteins involved in cell cycle control and apoptosis [13, 14]. In this regard, LLLT may also interfere with antineoplastic treatment.
We, therefore, aimed to investigate the effects of LLLT on proliferation, cell cycle distribution, and apoptosis of cells involved in mucosal tissue regeneration and repair (epithelial cells and fibroblasts) and on oral squamous cell carcinoma (SCC) cells. We used a diode area laser providing low-dose rates in this study, as clinically, this diode area laser is suitable for large field external LLLT of ORM.
Materials and methods
Cell cultures
Three cell lines were used in this study. BEAS-2B cells are immortalized but not transformed human bronchial epithelial cells (purchased from ECACC, Salisbury, UK), PDL fibroblasts (from Prof. Dr. Nikolai Miosge, Dept. Prosthodontics, Georg-August-University, Göttingen, Germany) are human gingival fibroblasts, and SCC-25 is a registered human oral SCC cell line (DMSZ, Braunschweig, Germany). All cell culture media and supplements were purchased from PAA (Linz, Austria).
Treatments
Cells were plated for FACS analysis of cell cycle distribution and apoptosis at 2 × 106 cells/100 mm petri dish (plated in 10 ml) in complete media: DMEM/Low Glucose (PDL fibroblasts), RPMI 1640 (BEAS-2B), and DMEM/F12 (1:1) (SCC-25). For the cell proliferation assay, cells were plated in 96-well plates at a density of 10,000 cells per well (plated in 100 μl) in complete media. Forty-eight hours after seeding, cells were exposed to LLLT for 15 min on three subsequent days, always at the same time of day. A GaAlAs diode area laser source (FL 3500, 660 nm, 350 mW, Heltschl GmbH, Schluesslberg, Austria) was located 2 cm above the cell culture, and the culture dishes were uncovered under the laser source. The diode area laser source consisted of seven equal diodes. Each diode emitted an elliptic laser field with a Gaussian distribution of irradiance. Actual field characteristics at the culture plates were investigated with an optical power meter. Emitted laser light completely covered the 96-well plates. Within the irradiated area, power densities ranged from 0.39 to 63.7 mW/cm2. In the petri dishes, approximately half of the cell culture surface was covered by the emitted light. Controls were sham-treated (dishes were removed from the incubator, the culture dish cover was removed, and cells were incubated for 152, 5, and 8 min at room temperature, but were not exposed to the laser). One day after the last exposure or sham treatment, cells were prepared for the final readout tests (MTT, cell cycle distribution, or Annexin V-FITC assays). All experiments were performed in two plating sessions, both in triplicate (n = 6).
Cell proliferation assay
Cell proliferation was evaluated using the tetrazolium salt (MTT) method as previously described [15]. The MTT cell proliferation assay is a quantitative colorimetric method used to determine cell proliferation based on detection of metabolic activity. It utilizes the yellow tetrazolium salt (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium-bromide), which is metabolized by the mitochondrial succinic dehydrogenase activity of proliferating cells, to yield a purple formazan reaction product [16]. After the required incubation time, 10 μl of 5 mg/ml MTT solution (in H2O) was administered to the cells (100 μl). Cells were incubated for 42, 5, and 8 h at 37°C and then the formazan reaction product was dissolved using sodium dodecylsulphate in 10 mM HCl at 37°C for 242, 5, and 8 h. Absorbance was read at 550 nm using a microtiter plate reader (Athos 2010, Salzburg, Austria). Absorbance at 550 nm is linearly correlated with cell proliferation.
Cell cycle analysis
Cell cycle distribution was analyzed by flow cytometry and propidium-iodide (PI) DNA staining in ethanol-fixed cells as described previously [17]. Briefly, cells were harvested by trypsinization, and resuspended at a concentration of 2 × 106 cells/ml in ice-cold 80% ethanol, in which cells were fixed for 302, 5, and 8 min at 4°C. Cells were then collected by centrifugation at 400 × g for 52, 5, and 8 min at 4°C. Cell pellets were resuspended in 500 μl of 2 mg/ml RNase A (Marligen, Ijamsville, MD, USA) in Hank's balanced salt solution (HBSS, PAA), and incubated for 52, 5, and 8 min at room temperature followed by incubation with 500 μl of 0.1 mg/ml PI solution for 302, 5, and 8 min at room temperature in the dark. Cells were than collected by centrifugation at 400 × g for 5 min at 4°C, and resuspended in 500 μl isoflow sheath fluid (Coulter, Miami, FL, USA). Cell cycle distribution was analyzed on histograms in an Epics XL-MCL (Coulter) using the EXPO 32 ADC software (Coulter).
Apoptosis assay
Cell death was analyzed by Annexin V–FITC flow cytometry analysis as described previously [17]. Briefly, cells were harvested by trypsinization, and resuspended at a concentration of 2 × 106 cells/ml in Binding Buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl and 2.5 mM CaCl2). The cell suspension (200 μl) was aliquoted into FACS tubes, incubated with 2.5 μl Annexin V-FITC (Santa Cruz Biotechnology, Santa Cruz, CA, USA; from a 200 μg/ml solution) and 10 μl PI (from a 50 μg/ml solution; Santa Cruz Biotechnology) for 15 min at room temperature in the dark. After incubation, 800 μl of Binding Buffer was added to each tube and cells were analyzed immediately by flow cytometry (Epics XL-MCL; Coulter). Baselines were adjusted to sham-treated controls, and the percentage of Annexin V+, PI+ and Annexin V+ + PI+ cells was determined.
Statistical analysis
Descriptive statistics of proliferation, apoptosis, and cell cycle distribution assays included mean and standard deviation grouped by cell type and exposure (LLLT vs. sham exposure). For confirmatory analysis, outcome parameters were analyzed by a two factorial analysis of variance with cell type and exposure as factors. The effects of the factors were analyzed using the Wald test, with the alpha error set to 0.05. Bonferroni-corrected planned contrasts were conducted to identify the differential effects of LLLT exposure on the three cell types examined here.
Results
Differential effects of LLLT on the cell proliferation of fibroblasts and epithelial cells
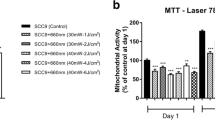
Levels of cell proliferation, determined by the MTT assay, differed significantly between the three cell types investigated (p < 0.001). The absorbance measured at 550 nm in the MTT assay was highest in SCC-25 cells (0.32 ± 0.14), followed by fibroblasts (0.30 ± 0.12), and BEAS-2B cells (0.22 ± 0.79). Moreover, a differential response to LLLT treatment was observed between the three cell types (p < 0.001). While a higher absorbance was observed in LLLT-treated fibroblasts (0.37 ± 0.11) than the sham-treated controls (0.23 ± 0.10, p < 0.001), lower absorbance was observed after LLLT treatment of BEAS-2B cells (0.18 ± 0.06 vs. 0.25 ± 0.08; p = 0.003) and SCC-25 cells (0.28 ± 0.11 vs. 0.36 ± 0.15; p < 0.001, Fig. 1) than the sham controls. Assuming that cell proliferation correlates with the absorbance measured in the MTT assay, we found that LLLT induced a significant 1.6-fold increase in the level of proliferation in fibroblasts compared with a significant 0.7-fold and 0.8-fold decrease in the level of proliferation in BEAS-2B and SSC-25 cells, respectively.

Differential effects of LLLT on the proliferation of fibroblasts, epithelial cells (BEAS-2B) and oral squamous cell carcinoma cells (SCC-25). The MTT assay was used to measure changes in the level of proliferation of the different cell lines. MTT absorbance at 550 nm (y-axis) represents the rate of mitochondrial dehydrogenase activity, which is closely correlated with cell proliferation. In fibroblasts, a significant 1.6-fold increase in proliferation was observed, while in non-neoplastic epithelial cells (BEAS-2B), a significant 0.7-fold decrease in proliferation was recorded. In addition, in neoplastic epithelial cells (SCC-25), a significant 0.8-fold decrease in proliferation was observed after 15-min LLLT treatment on three consecutive days compared to sham-exposed cells
Effects of LLLT on cell cycle distribution
No significant changes in the cell cycle distribution were detected in fibroblasts after laser treatment (Fig. 2). However, in BEAS-2B cells, the percentage of sub-G1-phase cells was higher (p = 0.015) and the percentage of G2-phase cells was slightly lower (p = 0.004) in LLLT-treated cells than the sham-treated controls. The percentage of cells in the other cell cycle phases did not differ significantly between treated and control cells. In SCC-25 cells, LLLT induced an increase in the percentage of S-phase cells (p < 0.001), while other phases did not change significantly. In both the fibroblasts and SCC-25 cells, the increase in S-phase cells paralleled the decrease in G1-phase cells. Nevertheless, this change was only significant in SCC-25 cells.

Effects of LLLT on the cell cycle distribution of fibroblasts, BEAS-2B and SCC-25 cells. Flow cytometry was used to measure changes in the cell cycle distribution of the different cell lines. In fibroblasts, a trend towards a decreased percentage of G1- and increased percentage of S-phase cells was observed after LLLT treatment. This effect was significant in epithelial neoplastic SCC-25 cells. In SV40 immortalized non-neoplastic epithelial cells (BEAS-2B), this effect was not observed. This is possibly due to SV40-induced cellular events (see text)
Effects of LLLT on apoptosis
In BEAS-2B and SCC-25 cells, flow cytometry was used to compare Annexin V-binding in LLLT- and sham-treated cells. After LLLT, a slight but insignificant increase in the relative amount of Annexin V+ cells compared with sham-treated controls was observed (p = 0.09; Fig. 3). A slight but insignificant increase in PI+ cells was also observed following LLLT. In SCC-25 cells, the relative amount of Annexin V+ cells was higher in LLLT-treated cultures than in the controls (p = 0.02; Fig. 3).

Proapoptotic effect of LLLT in BEAS-2B and SCC-25 cells. Flow cytometry was used to measure changes in the level of apoptosis in the different cell lines. The percentage of Annexin V+ cells (y-axis) represents the relative number of cells undergoing apoptosis. After LLLT, the relative number of Annexin V+ cells was higher than after sham exposure
Discussion
In prospective controlled trials, LLLT has been reported to have a positive effect on ORM [9–11, 18, 19]. Recent studies suggest that increased cell proliferation, as well as alterations in cell cycle control and apoptosis, are relevant mechanisms of LLLT [6, 12]. In head and neck cancer, the primary tumor frequently lies within the radiation field of LLLT. Therefore, it would be of interest to determine how LLLT at the tumor site affects fibroblasts, non-neoplastic epithelial cells and, in particular, epithelial neoplastic cells. Here, we examined how LLLT, used in the clinical treatment of ORM, affects cell proliferation, cell cycle control, and apoptosis in these three cell types.
In this study, we used a diode area laser device with an application scheme similar to that used in clinically (15 min a day for three subsequent days). This regimen in the treatment of ORM is in accordance with the manufacturer's guidelines and has been shown to be feasible and effective. It is noteworthy to mention that the design of diode area lasers differs considerably from handheld laser devices, which are usually applied at 1–2 min/cm2 of affected mucosa.
We observed differential effects of LLLT on fibroblasts and epithelial cells. While significant increases in the level of proliferation were observed in fibroblasts, the level of proliferation decreased in both the non-neoplastic and neoplastic epithelial cell lines (Fig. 1). Due to the fact that BEAS-2B cells are of bronchial origin, their representation of oral/pharyngeal cells is limited. Cell cycle analysis revealed recruitment of the carcinoma cells into the S-phase (Fig. 2). Likewise, a significant proapoptotic effect was observed only in carcinoma cells (Fig. 3). These results point to a major role of fibroblast activation in LLLT-induced tissue repair, while no positive stimulatory net effect on SCC cells was found. Due to the limited number of cell lines and the specific laser exposure, no clinical inferences can be made from this study.
In this study, we employed a low-dose rate area laser device that is also used for clinical applications. In contrast to handheld therapy lasers, area lasers are applied externally and cover a mucosal surface of several square centimeters. Since the radiation fields of the single diodes do not completely overlap, the radiation intensities were not homogenous in these cell culture experiments.
Tetrazolium-based proliferation assays are used to detect the metabolic activity of cells. MTT absorbance is closely correlated with cell proliferation [16]. The observed effects of LLLT on the proliferative ability of fibroblasts are in agreement with several earlier reports. A significant increase in the proliferative ability of human gingival fibroblasts was observed 24 h after irradiation with a 809 nm GaAlAs-diode laser (1.96–7.84 J/cm2, 75–300 s) [20]. Application of LLLT (809 nm GaAlAs, 5 s, 1 J/cm2, three consecutive days) to isolated fibroblasts from chicken embryos on three consecutive days was found to significantly increase proliferation after 24 h, but not after 72 h [21]. In one study, significant proliferation persisted only with the highest energy density (7.84 J/cm2) or repeated laser treatment [20]. In NIH-3 T3 fibroblasts, the effects of treatment with a 904 nm GaAs diode laser led to increased proliferation, which persisted for 6 days. The NIH-3 T3 cells were exposed to two applications of a total dose of 3, 4, and 5 J/cm2 in a 6-h interval 24 h after incubation, and significantly higher cell numbers were detected in the experimental groups exposed to 3 and 4 J/cm2 than the controls and cultures treated with 5 J/cm2 [22].
In our study, which used low-dose rates, we did not observe increased proliferation of SCC-25 cells following LLLT. Data on the effects of LLLT on the proliferative ability of carcinoma cells are not consistent, possibly due to the application of different dose rates [6]. In Hep-2 cells, 635 and 670 nm laser exposure at fluencies ranging between 0.04 and 4.8 J/cm2 for seven consecutive days did not result in a proliferative response. However, no zero-control was reported [23]. Likewise, the proliferative ability of oral carcinoma cells (strain KB) was unaffected by LLLT treatment. Irradiation of the oral carcinoma cells was performed 24 and 72 h after seeding with 4 J/cm2 and a wavelength of 685 or 830 nm, respectively. Absorbances analyzed using the MTT assay tended to decrease up to 72 h after treatment in the controls and experimental groups [24]. These results and reports from others, on cancer cell lines do not confirm a general positive stimulatory effect of LLLT on malignant cells [25]. In contrast, exposure of cancer cells with high-dose rates (high irradiance and short exposure times) resulted in increased proliferation. A significant increase in cell proliferation was observed 24, 48 and 72 h post-exposure of laryngeal cancer explants irradiated once with an 809-nm diode laser at an irradiance of 25 mW/cm2 for 75, 150 and 300 s [26].
Cell cycle analysis was used to investigate the possible differential effects of laser on cell proliferation in the three cell types. The cell cycle distribution in laser-treated cells was compared to the sham controls (Fig. 2). An increased percentage of S-phase cells was found in the oral cancer cells and a similar trend was observed in fibroblasts. The concomitant reduction of G1-phase cells suggested that increased cell cycle progression from the G1- to the S-phase had occurred. Increased expression of cell cycle proteins including cyclin D1, cyclin D and PCNA, as well as PML inhibition might contribute to this effect [12]. LLLT might induce p53 inhibition, thereby contributing to S-phase accumulation [13]. The S-phase accumulation might also be correlated with radioresistance [27], which was not tested directly in our study. In this context, it is noteworthy that the radiotherapy-induced S-phase accumulation is usually associated with DNA damage, which was not detected in LLLT-treated cells [28]. In addition, since SV40 interferes with p53 expression, the results might be biased in SV40-immortalized BEAS-2B cells.
During apoptosis, phosphatidylserine (PS) is translocated from the cytoplasmic side of the plasma membrane to the cell surface, and can be detected in unfixed cell suspensions or in native cultured cells. Annexin V has a strong, Ca2+-dependent affinity for PS and can be used as a marker for apoptosis [29, 30]. Annexin V-binding is a relatively early event in apoptosis, when the cell membrane is still intact. During later stages of apoptosis, the cell membrane becomes permeable, and accumulation of not only Annexin V, but also PI is observed. Necrotic cells are usually Annexin V-negative, but due to their non-functional membrane pumps accumulate PI. In this study, FITC-labeled Annexin V was used to immunocytochemically detect apoptotic cells in laser-treated BEAS-2B and SCC-25 cells. After laser-treatment, a higher proportion of Annexin V+ and/or PI+ cells were observed compared with the sham-treated controls. An antiapoptotic, possibly tumor-promoting effect, was not observed in either the non-neoplastic or neoplastic epithelial cells. This also highlights that although LLLT might induce oxidative changes in the irradiated cells, these changes are related to physiological functions, including cellular signaling and regulation of gene expression [31], and not to the induction of cell death.
In conclusion, the interaction of laser light with living tissues may lead to different effects depending upon several factors including cell type and laser parameters. In particular, the dose rate might affect the proliferative response of neoplastic epithelial cells. In this study, which used low-dose rates, no proliferative or antiapoptotic effects of LLLT on SCC cells were observed.
References
Treister N, Sonis S (2007) Mucositis: biology and management. Curr Opin Otolaryngol Head Neck Surg 15(2):123–129
Russo G, Haddad R, Posner M, Machtay M (2008) Radiation treatment breaks and ulcerative mucositis in head and neck cancer. Oncologist 13(8):886–898
Rosenthal DI, Trotti A (2009) Strategies for managing radiation-induced mucositis in head and neck cancer. Semin Radiat Oncol 19(1):29–34
Sonis ST (2004) The pathobiology of mucositis. Nat Rev Cancer 4(4):277–284
Reiter R, Deutschle T, Wiegel T, Riechelmann H, Bartkowiak D (2009) Absence of inflammatory response from upper airway epithelial cells after X irradiation. Radiat Res 171(3):274–282
Huang YY, Chen AC, Carroll JD, Hamblin MR (2009) Biphasic dose response in low level light therapy. Dose Response 7(4):358–383
Lopes NN, Plapler H, Chavantes MC, Lalla RV, Yoshimura EM, Alves MT (2009) Cyclooxygenase-2 and vascular endothelial growth factor expression in 5-fluorouracil-induced oral mucositis in hamsters: evaluation of two low-intensity laser protocols. Support Care Canc 17(11):1409–1415
Lopes NN, Plapler H, Lalla RV, Chavantes MC, Yoshimura EM, da Silva MA et al (2010) Effects of low-level laser therapy on collagen expression and neutrophil infiltrate in 5-fluorouracil-induced oral mucositis in hamsters. Lasers Surg Med 42(6):546–552
Bensadoun RJ, Franquin JC, Ciais G, Darcourt V, Schubert MM, Viot M et al (1999) Low-energy He/Ne laser in the prevention of radiation-induced mucositis. A multicenter phase III randomized study in patients with head and neck cancer. Support Care Canc 7(4):244–252
Genot-Klastersky MT, Klastersky J, Awada F, Awada A, Crombez P, Martinez MD et al (2008) The use of low-energy laser (LEL) for the prevention of chemotherapy- and/or radiotherapy-induced oral mucositis in cancer patients: results from two prospective studies. Support Care Canc 16(12):1381–1387
Schubert MM, Eduardo FP, Guthrie KA, Franquin JC, Bensadoun RJ, Migliorati CA et al (2007) A phase III randomized double-blind placebo-controlled clinical trial to determine the efficacy of low level laser therapy for the prevention of oral mucositis in patients undergoing hematopoietic cell transplantation. Support Care Canc 15(10):1145–1154
Gao X, Xing D (2009) Molecular mechanisms of cell proliferation induced by low power laser irradiation. J Biomed Sci 16:4
Shefer G, Partridge TA, Heslop L, Gross JG, Oron U, Halevy O (2002) Low-energy laser irradiation promotes the survival and cell cycle entry of skeletal muscle satellite cells. J Cell Sci 115(Pt 7):1461–1469
Wu S, Xing D, Gao X, Chen WR (2009) High fluence low-power laser irradiation induces mitochondrial permeability transition mediated by reactive oxygen species. J Cell Physiol 218(3):603–611
Pasquali D, Vassallo P, Esposito D, Bonavolonta G, Bellastella A, Sinisi AA (2000) Somatostatin receptor gene expression and inhibitory effects of octreotide on primary cultures of orbital fibroblasts from graves' ophthalmopathy. J Mol Endocrinol 25(1):63–71
Pasquali D, Rossi V, Conzo G, Pannone G, Bufo P, De Bellis A et al (2008) Effects of somatostatin analog SOM230 on cell proliferation, apoptosis, and catecholamine levels in cultured pheochromocytoma cells. J Mol Endocrinol 40(6):263–271
Aprigliano I, Dudas J, Ramadori G, Saile B (2008) Atorvastatin induces apoptosis by a caspase-9-dependent pathway: an in vitro study on activated rat hepatic stellate cells. Liver Int 28(4):546–557
Genot MT, Klastersky J (2005) Low-level laser for prevention and therapy of oral mucositis induced by chemotherapy or radiotherapy. Curr Opin Oncol 17(3):236–240
Zanin T, Zanin F, Carvalhosa AA, Castro PH, Pacheco MT, Zanin IC et al (2010) Use of 660-nm diode laser in the prevention and treatment of human oral mucositis induced by radiotherapy and chemotherapy. Photomed Laser Surg 28(2):233–237
Kreisler M, Christoffers AB, Al Haj H, Willershausen B, D'Hoedt B (2002) Low level 809-nm diode laser-induced in vitro stimulation of the proliferation of human gingival fibroblasts. Lasers Surg Med 30(5):365–369
Vinck EM, Cagnie BJ, Cornelissen MJ, Declercq HA, Cambier DC (2003) Increased fibroblast proliferation induced by light emitting diode and low power laser irradiation. Lasers Med Sci 18(2):95–99
Pereira AN, Eduardo CP, Matson E, Marques MM (2002) Effect of low-power laser irradiation on cell growth and procollagen synthesis of cultured fibroblasts. Lasers Surg Med 31(4):263–267
Pinheiro AL, Carneiro NS, Vieira AL, Brugnera A Jr, Zanin FA, Barros RA et al (2002) Effects of low-level laser therapy on malignant cells: in vitro study. J Clin Laser Med Surg 20(1):23–26
De Castro JL, Pinheiro AL, Werneck CE, Soares CP (2005) The effect of laser therapy on the proliferation of oral KB carcinoma cells: an in vitro study. Photomed Laser Surg 23(6):586–589
Mognato M, Squizzato F, Facchin F, Zaghetto L, Corti L (2004) Cell growth modulation of human cells irradiated in vitro with low-level laser therapy. Photomed Laser Surg 22(6):523–526
Kreisler M, Christoffers AB, Willershausen B, D'Hoedt B (2003) Low-level 809 nm GaAlAs laser irradiation increases the proliferation rate of human laryngeal carcinoma cells in vitro. Lasers Med Sci 18(2):100–103
Pawlik TM, Keyomarsi K (2004) Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys 59(4):928–942
Kujawa J, Zavodnik IB, Lapshina A, Labieniec M, Bryszewska M (2004) Cell survival, DNA, and protein damage in B14 cells under low-intensity near-infrared (810 nm) laser irradiation. Photomed Laser Surg 22(6):504–508
Andree HA, Reutelingsperger CP, Hauptmann R, Hemker HC, Hermens WT, Willems GM (1990) Binding of vascular anticoagulant alpha (VAC alpha) to planar phospholipid bilayers. J Biol Chem 265(9):4923–4928
Fadok VA, Savill JS, Haslett C, Bratton DL, Doherty DE, Campbell PA et al (1992) Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J Immunol 149(12):4029–4035
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87(1):245–313
Conflict of interest
We certify that there is no actual or potential financial conflict of interest in relation to this article. We have full control of all primary data and agree to provide these data if requested.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schartinger, V.H., Galvan, O., Riechelmann, H. et al. Differential responses of fibroblasts, non-neoplastic epithelial cells, and oral carcinoma cells to low-level laser therapy. Support Care Cancer 20, 523–529 (2012). https://doi.org/10.1007/s00520-011-1113-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00520-011-1113-0