
Abstract
This study was designed to characterize extended-spectrum beta-lactamase (ESBL)–producing extra-intestinal pathogenic Escherichia coli (E.coli) (ExPEC) associated with urinary tract infections in nine different geographic regions of Zimbabwe over a 2-year period (2017–2019). A total of 48 ESBL-positive isolates from urine specimen were selected for whole-genome sequencing from 1246 Escherichia coli isolates biobanked at the National Microbiology Reference laboratory using phenotypic susceptibility testing results from the National Escherichia coli Surveillance Programme to provide representation of different geographical regions and year of isolation. The majority of ESBL E. coli isolates produced cefotaximase-Munich (CTX-M)-15, CTX-M-27, and CTX-M-14. In this study, sequence types (ST) 131 and ST410 were the most predominant antimicrobial-resistant clones and responsible for the increase in ESBL–producing E. coli strains since 2017. Novel ST131 complex strains were recorded during the period 2017 to 2018, thus showing the establishment and evolution of this antimicrobial-resistant ESBL clone in Zimbabwe posing an important public health threat. Incompatibility group F plasmids were predominant among ST131 and ST410 isolates with the following replicons recorded most frequently: F1:A2:B20 (9/19, 47%), F2:A1: B (5/19, 26%), and F1:A1:B49 (8/13, 62%). The results indicate the need for continuous tracking of different ESBL ExPEC clones on a global scale, while targeting specific STs (e.g. ST131 and ST410) through control programs will substantially decrease the spread of ESBLs among ExPEC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The production of extended-spectrum beta-lactamases (ESBLs) by clinical isolates of extra-intestinal pathogenic Escherichia coli (ExPEC) is a serious global therapeutic threat [1]. These ESBLs can reduce the efficacy of the extended-spectrum cephalosporins, except for cephamycin and carbapenems [2]. ESBL production is associated with the presence of blaTEM, blaSHV, and blaCTX-M [3]. CTX-M producers are increasingly detected and replacing TEM and SHV producers in European and African countries [4]. A number of important E. coli clones have been detected among the ESBL producers, these include sequence type (ST) 131, ST405, ST38, ST648, ST410, and ST1193 [1].
Among these, the most studied phylogenetic lineage of E. coli in terms of antibiotic resistance is E. coli ST131, phylogenetic group B2 serotype O25:H4. This lineage harbours a wide range of core sets of virulence genes and various plasmid-mediated resistance genes. ST131 is involved in the global spread of the ESBL phenotype linked to the production of CTX-M-15 and CTX-M-27, which is the main mechanism of resistance to beta-lactams [1]. ST131’s virulence combined with its carriage of transferable elements encoding multidrug resistance is likely responsible for the pandemic success of ST131 strains [5, 6].
The ST131 population structure is divided into three clades, namely, A, B, and C. The C clade comprises two subclades, C1 and C2, which are defined by the presence of a specific fimbrial adhesin allele (fimH30) corresponding to the H30R and H30Rx clades [7]. The C1 subclade contains mutations in the chromosomal genes gyrA and parC that confer fluoroquinolone resistance, while the C2 subclade contains the same gyrA and parC mutations but is also associated with blaCTX-M-15. Reports of the occurrence of ST131 subgroup C1-M-27 associated with blaCTX-M-27 in some parts of the world especially in Europe [8], Asia, and parts of Africa [9] emerged.
ESBL–producing E. coli associated with nosocomial- and community-acquired infections have been reported in most regions around the world [10]. These bacteria are resistant to most of the antimicrobials used for the treatment of urinary tract infections (UTIs), such as ciprofloxacin, trimethoprim-sulfamethoxazole, and most of the cephalosporins [11]. A few studies have been carried out in sub-Saharan Africa (South Africa, Nigeria, Tanzania, and Democratic Republic of Congo) to investigate the molecular epidemiology and characteristics of the ExPEC strains [12,13,14,15], while in Zimbabwe, this remained unexplored. Thus, the main aim of this study was to characterize the ESBL ExPEC associated with UTIs in different geographic regions of Zimbabwe using whole-genome sequencing (WGS).
Materials and methods
Selection of bacterial isolates for genomic evaluation
A total of 48 ESBL-positive isolates from urine specimens were selected for WGS from 1246 E. coli isolates biobanked at the National Microbiology Reference laboratory using phenotypic susceptibility testing results from the National Escherichia coli Surveillance Programme to provide representation of different geographical regions and years of isolation (2017 (14); 2018 (22), and 2019 (12)) (Supplementary File 1Epidemiological features of the 48 ESBL isolates). Demographic data associated with 48 ESBL-positive isolates were analyzed. The geographical regions included Bulawayo, Chitungwiza, Marondera, and Harare. The selection was also based on community-acquired UTIs, which was defined as an infection of the urinary tract that occurs in the community or within 48 h of hospital admission and was incubating during time of hospital admission [16]. No information of previous use of antibiotics was collected.
Study population phenotypic antibiotic resistance profiling
All the selected isolates were sub-cultured on MacConkey or eosin methylene blue (EMB) (Mast Group, Merseyside, UK), incubated in Memmert ICH110 (Germany) at 37 °C for 18 to 24 h and then stored in 20% glycerol broth at 80 °C. Antimicrobial susceptibility testing was determined by the Kirby Bauer disc diffusion method using the Clinical Laboratory Standards Institute (CLSI) guidelines [17]. The antimicrobial drugs tested included ampicillin, trimethoprim-sulfamethoxazole, ciprofloxacin, ceftriaxone, tetracycline, ceftazidime, nalidixic acid, cefepime, and ertapenem; results were interpreted as described by the CLSI [17]. The presence of ESBLs was confirmed according to the CLSI criteria for ESBL screening and confirmation [17]. E. coli ATCC 25922 was used as quality control strain. Additional data included on collection of each isolate were the year, location of isolation, travel associated, cities, age, and gender (Supplementary File 1.
Genomic DNA isolation and whole-genome sequencing
Genomic DNA (gDNA) of E. coli was purified using the Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions [18] and stored at – 20 °C. Library preparation was performed using the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) and sequenced on a MiSeq benchtop sequencer (Illumina, San Diego, CA, USA) at Quadram Institute Biosciences in the UK. The data was uploaded to BaseSpace (http://www.basespace.illumina.com) and then converted to FASTQ files.
Genomic sequence analysis
The sequences were analyzed on the Cloud Infrastructure for Microbial Bioinformatics [19]. Paired-end short-read sequences were concatenated, then quality-checked using FastQC v0.11.7. De novo assembly was performed with SPAdes 3.11 [20], and quality was assessed using QUAST 4.5 [21]. Snippy v4.3.2 (https;//github.com/tseemann/snippy) was used to generate a core SNP alignment using default parameters. The complete genome sequence of E. coli strain K12 sub-strain MG1655 was used as reference genome (NCBI accession: NC_000913.3). After the core-genome alignment, a reconstruction of maximum likelihood phylogeny with 1000 bootstrap replicates with RAxML v8.2.4 based on a general time-reversible nucleotide substitution model was used [22, 23]. The phylogenetic tree was rooted using Escherichia fergusonii (E. fergusonii) as an outgroup (NCBI accession: GCA_000026225.1). The phylogenetic tree was visualized in Figtree v1.4.4 (https://github.com/rambaut/figtree/) [24] and annotated in RStudio v3.5.2 and Adobe illustrator v 23.0.3 (Adobe Inc. San Jose, CA). Recombination was detected and masked using Gubbins [25] before the phylogenetic reconstruction. The pairwise SNP distances were computed between genomes from the core-genome alignment using snp-dists v0.6 (https://github.com/tseemann/snp-dists).
Comparative genomics analysis
The assembled draft genomes were used to define the presence of genes and their alleles. The following databases or typing schemes were used to determine (i) STs (multi-locus sequence typing (MLST) method according to Achtman scheme, https://guthub.com/tseemann/mlst) [26]; (ii) phylogenetic groups (ClermonTyper v1.0.0) [27]; (iii) resistance genes (ARIBA database) [28]; (iv) virulence factors (virulence factor database, VFDB) [29]; (v) serotypes (serotypeFinder O:H typing database) [30]; (vi) plasmids (PlasmidFinder) [26]; and (vii) sequence types for plasmids (plasmidMLST) [26] (Supplementary File 1). Novel STs were assigned by Enterobase [26].
A phylogenetic tree was constructed using recombination-free core genomes including ST131 genomes from Africa retrieved from Enterobase, representative clade strains from Matsumura et al. [9] and this study’s ST131 sequence data to demonstrate the phylogenetic relatedness of isolates. The isolates EC958 fimH30 and KUN2145 fimH22 were used as reference and an outgroup, respectively. A similar comparative analysis was done for this study’s ST410 isolates in comparison to ST410 genomes from Africa submitted to Enterobase, Roer et al. [31], and this study’s sequence data. The reference genome used for the phylogenetic tree construction was YD786 (GenBank Accession Number: NZ_CP013112.1), while fimH53 isolates were used as outgroups for the comparative analysis of ST410 isolates. Only ST131 and ST410 genomic data with relevant metadata (year of collection, country, source type, etc.) and availability of raw reads on Enterobase were included. Mauve was used to visualize similarities of this study’s ST131 C1-M27 genomic environments against a reference KUN 5781 for the presence of a phage integrase region annotated as M27PP1 and M27PP2.
Ethical approvals
Ethical clearance was obtained from the Faculty of Health Sciences Research Ethics Committee, University of Pretoria (Ethics Reference Number: 782/2018) and the Medical Research Council of Zimbabwe, Approval Number: MRCZ/A/2394.
Results
Baseline characteristics of sequenced isolates
A total of 48 ESBL-positive isolates from three provinces (Harare, Bulawayo, and Mashonaland East) were analyzed. Among these, 27 (55%) were linked to UTIs in female patients and 21 (45%) to male patients. Thirty (62%) patients were aged 21 to 60 years. Thirty-eight (80%) of the isolates were recovered from Harare with isolation years (2017 (11); 2018 (16); 2019 (11)), while five isolates (10%) originated from Chitungwiza (2017 (2); 2018 (3)), three isolates (6%) were collected from Bulawayo (2018 (2); 2019 (1)), and two (4%) were from Marondera (2017 (1); 2018 (1)). These ESBL-producing ExPEC isolates originating from the outpatient clinics of hospitals in Harare, Chitungwiza, Marondera, and Bulawayo displayed increased resistance to all antimicrobials included in the study.
Whole-genome sequence analysis
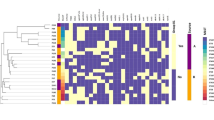
The phylogenetic tree in Fig. 1 was constructed using representative reference strains of phylogenetic groups A, B1, B2, D, and E and an outgroup E. fergusoni. The isolates in this study were compared in terms of the virulence factors detected and those isolates belonging to the same phylogenetic group as the representative reference strains clustered together as shown in Fig. 1.

A maximum likelihood phylogeny of the study isolates reconstructed with RAxML, based on non-repetitive, non-recombinant core SNPs using a general time-reversible nucleotide substitution model with 1000 bootstrap replicates. The top labels indicate sample names with respective phylogroups and STs to which the isolates belong. The outgroup (not shown) and the other E. coli reference genomes denoting the major E. coli phylogroups are in bold italics (A (MG1655), B1 (IAI1), B2 (CFT073), D (UMN026), and E (EDL933)). Overlaid on the tree are the years of sample isolation, Zimbabwe locations, and virulence factors for each isolate. The virulence genes are grouped according to their functions
Genomic assembly, quality control, and phylogenetic tree
Forty-eight isolates had a combined length of contigs of assembled genomes ranging from ~ 4.7 to 9.8 Mbp, with a minimum contig length required to cover 50% of the genome (N50), ranging between 84 and 340 kbp. The SNP matrix output tables with sequenced isolates aligned and compared to reference genomes were used to construct a phylogenetic tree as shown in Fig. 1.
Beta-lactamases, plasmid-mediated quinolone resistance determinants, and phylogenetic groups
The 48 ESBL E. coli isolates harboured diverse ESBL genes; blaCTX-M-15 (34), blaCTX-M-27 (11), blaCTX-M-14 (1), blaCTX-M-3 (1), and blaCTX-M-82 (1). Other ESBL genes were also observed, including blaTEM-1, blaOXA-1, and blaOXA-10 (Table 1). Table 2 illustrates the different beta-lactamases that were detected in different geographic regions and years. Other antimicrobial resistance (AMR) genes included the aac (6′)-lb-cr gene (27) encoding aminoglycoside acetyltransferase, the qnrS gene (4) encoding a plasmid-mediated quinolone resistance, and the qepA (1) encoding a quinolone pump. Twenty isolates belonged to phylogenetic group B2; 14 to phylogenetic group C; four to phylogenetic group B1; three to phylogenetic groups A, D, and F each; and lastly one to phylogenetic group G.
Multi-locus sequence typing
MLST identified five major ST clonal complexes (CCs), which included the following: ST131CC as ST131 (17), ST11380 (1), and ST11387 (1); ST23CC as ST410 (13) and ST6332 (1); ST10CC as ST617 (2) and ST218 (1); ST405CC as ST405 (1) and ST11615 (1); ST648CC as ST648 (1); ST354CC as ST354 (1); ST155CC as ST155 (1). Most of the isolates (43/48) belonged to one of these defined CCs (Table 1). Five isolates belonged to an undefined clonal complex, referred to as “unknown CC”, which included ST2448 (1), ST117 (1), ST224 (1), ST678 (1), ST636 (1). The clinical presentation, year of collection, antimicrobial susceptibility profile, plasmid-mediated quinolone-resistant (PMQR) determinants, ESBL types, and phylogenetic groups of the above defined and undefined CCs are shown in Tables 1, 2, and Supplementary File 1.
Plasmid replicon types
The most predominant plasmids were Col156, ColBS512, and ColMG828, which were distributed among the ST131 and ST410 isolates as shown in Table 3. All ST131 and ST410 isolates characterized in this study harboured incompatibility group F1A (IncF1A) plasmids. Different plasmid types belonging to IncF were distributed across all strains. The IncF plasmids with F1:A2:B20 (9/19) and F2:A1: B (5/19) replicons were predominant among ST131 isolates, while the F1:A1:B49 replicon (8/14) predominated among the ST410 strains. The F36:A4: B1 replicon was detected among isolates from ST23, ST131, and ST10 CCs as shown in Supplementary File 1.
E. coli ST131 core genome SNP-based phylogenetic tree in an African and global context
The mapping and alignment of 19 ST131 study isolates to the reference genome EC958 produced a core genome of 4,700,950 bp. The fluoroquinolone-resistant isolates with gyrA and parC mutations formed the C/H30R cluster that comprised the C2/H30Rx and C1/H30R clades. The C2/H30Rx clade included isolates with blaCTX-M-15 (n = 43) and isolates with both blaCTX-M-14 and blaCTX-M-15 (n = 1), and blaCTX-M-15 and blaCTX-M-27 (n = 1) and isolates without blaCTX-M (n = 2). The C1/H30R clade included isolates with blaCTX-M-27 (n = 17) and blaCTX-M-14 (n = 8) and isolates without ESBLs (n = 1) as shown in Fig. 2. Most Zimbabwean isolates belonged to the C2/H30Rx clade (10/19), with between 10 and 15 SNP differences between isolates from neighbouring African countries, such as Tanzania, DRC, Nigeria, Sudan, Niger, and Ethiopia as shown in Supplementary File 2. Within the C1/H30R clade, 15 of the 17 CTX-M-27-producing isolates clustered into a distinct group, the C1-M27 clade. E. coli ST131 C1-M27 comprised isolates from Zimbabwe (n = 9, 2017–2019); Canada (n = 1, 2008); USA (n = 2, 2013–2014); Japan (n = 2 2007, 2010); and Australia (n = 1, 2009). The novel STs assigned by Enterobase in the ST131 CC clustered in the C1-M27 clade, and these were closely related to Canadian strains (Fig. 2).

Core genome single-nucleotide polymorphism (SNP)–based phylogenetic tree of Escherichia coli sequence type 131 comparison of isolates from Africa uploaded on Enterobase, Europe [9], and this study. The maximum likelihood phylogenetic tree is based on a 4,720,950-bp core genome and 5000 SNPs. The tree is rooted by using the outgroup H22 isolate. A general time-reversible nucleotide substitution model with 1000 bootstrap replicates was used. Strains from Matsumura et al. [9] are all italicized, those from Africa (Enterobase) are in bold, and this study’s isolates have been labelled with an asterisk
The C1-M27 clade-specific region
The genome analysis of C1-M27 clade isolates identified an 11,894-bp region that is named M27PP1, which is a prophage integrase specific to all isolates from the C1-M27 clade and to some non-ST131 from this study that had a CTX-M27 resistance gene. BLAST and Mauve software were used to align the isolates to a reference with these regions clearly annotated (KUN5781). Three isolates from C1-M27 clade (i.e. 58EC, 60EC, 92EC) and two non-ST131 isolates (i.e. 72EC, 69EC) from this study had M27PP1 alone. Six E. coli ST131 C1-M27 isolates (i.e. 38EC, 45EC, 51EC, 53EC, 91EC, 67EC) aligned against the reference KUN5781 as shown in Fig. 3 had an additional insertion region named the M27PP2 situated upstream of M27PP1.

Genetic environment of the C1-M27 clade-specific region of Escherichia coli
E. coli ST410 in Africa and global context
The phylogenetic reconstruction of 127 ST410 international isolates, 55 African isolates retrieved from Enterobase, and 17 isolates from Zimbabwe in this study revealed two distinct clonal lineages of ST410: lineage A with fimH53 (A/H53) and lineage B with fimH24 (B/H24) (Fig. 4). The B/H24 lineage was further divided into three sublineages: B2/H24R with the introduction of fluoroquinolone resistance by mutations in the gyrA and parC, B3/H24Rx with the introduction of blaCTX-M-15, and B4/H24RxC with the introduction of blaOXA-181. Zimbabwean isolates were all part of the B3/H24Rx clade.

Core genome single-nucleotide polymorphism (SNP)–based phylogenetic tree of Escherichia coli sequence type 410 comparison of isolates from Africa retrieved from Enterobase, Europe [31], and this study. The rooted maximum likelihood phylogenetic tree was constructed with Gubbins-based SNP alignment. The tree was rooted by using the outgroup fimH53 isolates. A general time-reversible nucleotide substitution model with 1000 bootstrap replicates was used. Strains from Roer et al. [31] are all italicized, those from Africa (Enterobase) are indicated in bold, and this study’s isolates have been labelled with an asterisk
Discussion
Emerging and established high-risk clones of ESBL-producing ExPEC are factors that negatively impact on human health management globally. This study focused on elucidating the molecular characteristics of ESBL–producing E. coli isolates associated with UTIs over a 2-year period (2017–2019) in Zimbabwean communities (Harare, Marondera, Bulawayo, and Chitungwiza). The Zimbabwean uropathogenic E. coli isolates harboured a high rate (28%) of ESBL production; similarly, high rates have been reported in studies from Tanzania (24%) [32], Algeria (31%) [33], and Rwanda (38%) [34]. In Zimbabwe’s hospital setting, beta-lactam antibiotics, such as ceftriaxone, are frequently used as first-line treatment for bacterial infections, which create a selective pressure for the pathogens to evolve and adapt [35, 36].
The high diversity of uropathogenic ESBL–producing E. coli isolates in this study suggests the circulation of diverse community-acquired UTI clones within Zimbabwe. Most isolates belonged to major CCs, such as ST131, ST23 (ST410), ST10, ST405, ST648, ST69, ST354, and ST155. The pandemic clone, ST131, was the most detected clone in this study and is known to harbour blaCTX-M-15, blaCTX-M-27, and fluoroquinolone resistance. The presence of this clone has been reported in small sample size studies from South Africa [37], Nigeria [38], and Tunisia [39]. This clone is known for its high virulence leading to infections, such as invasive bloodstream, urinary tract, and intra-abdominal infections [40].
A comparative analysis on the phylogenetic relatedness of this pandemic clone in relation to those reported from other African countries revealed a possible constant influx of ST131 strains as these isolates are mixed with other African and European sublineages in the phylogeny (Fig. 2). A local national transmission cluster of ST131 blaCTX-M-27 was observed. This clade named C1-M27 within C1/H30R in ST131 has been previously reported as responsible for epidemics of ESBL-producing ExPEC in Japan [9] and has so far disseminated across different continents including Africa. The isolates clustering in this clade originated from Harare and Marondera. The proximity of these two cities could have contributed to the spread of C1-M27, although information on travel could not be retrieved upon collection of samples. Local transmission could be the largest contributor to the spread of infections with ESBL–producing E. coli ST131 in Zimbabwe given the small number of SNPs between isolates in the C1-M27 clade. Travel history becomes an important characteristic to consider in future studies as a better method to link these transmission dynamics.
The Zimbabwean isolates are defined by the presence of either the M27PP1 unique region or by both the M27PP1 and M27PP2 insertion regions. The E. coli ST131 C1-M27 isolates harboured the unique prophage-like region (M27PP1) within its chromosome, while in ST131 C2/H30Rx, it was not identified (Fig. 3). The direct flanking repeat sequences surrounding M27PP1 suggest that this region was introduced into E. coli ST131 C1/H30R with blaCTX-M-27 by a recombination event that was then followed by the clonal expansion of the C1-M27 clade [8, 9]. Therefore, the screening for the presence of M27PP1 genetic environment is important to check for recombination since other studies have noticed that some ST131 isolates might have acquired blaCTX-M-27 independently from the C1-M27 clade [8, 9].
UTIs are often preceded by colonization of the gut [41]. In a recent study by Wilmore et al. [42] on the carriage of ESBL–producing Enterobacteriaceae in HIV-infected children in Zimbabwe, it was observed that out of 175 collected stools, 24 isolates were ESBL-positive and nine isolates belonged to ST131 producing either CTX-M-15 or CTX-M-27. Infection as a result of colonization by these resistant strains may complicate treatment. The presence of such high rates of ESBL–producing commensal bacteria is a reflection of the high usage of these antibiotics in the public sector in Zimbabwe and its contribution to the creation of selective pressure for pathogens to evolve and spread.
Novel strains of the ST131 complex (ST11380 and ST11387) and ST405 complex (ST11615) were detected over a period of 1-year (2017–2018) harbouring blaCTX-M-15, and blaCTX-M-27. These novel strains originated from patients who live in one of the densely populated suburbs, where frequently poor sanitation and hygiene conditions are reported. As these resistant clones can persist in the environment, it is important to improve sanitation, water quality, and patient care through education and awareness campaigns in communities as a way of controlling the spread of such resistant clones [43, 44]. From our global analysis, other environmental-associated isolates were included, which showed the existence of different clades including C1-M27 among animal and environment; this shows how such isolates might act as reservoirs for the introduction of such clades in humans.
Enterobacterales are known for having relatively open pan-genomes that can rapidly adapt to changing selection pressures (including antibiotic usage) as observed in the ST131 and ST410 strains in this study. To the best of the authors’ knowledge, this is the first report of ST410 ESBL ExPEC from Zimbabwe harbouring blaCTX-M-15 along with other antimicrobial resistance (AMR) genes. Recent studies have indicated the E. coli ST410 as another successful pandemic ExPEC lineage [31]. Our study results corroborate this theory; however, as national surveillance programmes monitoring only local epidemiology, global surveillance programmes are required to follow the dissemination of pandemic clones [45]. A comparative analysis of the ST131 and ST410 isolates revealed the presence/predominance of the self-transmissible IncF plasmids in ST410, which like in ST131 allows the bacteria to capture additional virulence genes and resistance determinants [5]. Similar studies on poultry, companion animals, freshwater fisheries, and swine from Tunisia and South America have described the presence of IncF plasmids in ST410 as a contributing factor to the spread of ESBL [46, 47]. Due to the lack of ecological barriers and trading of food items between nations and continents, feacal carriage of these resistant clones may contribute greatly to the spread of AMR [48].
IncF-type plasmids have a narrow host range (limited to Enterobacteriaceae) and contribute to bacterial fitness via antibiotic resistance and virulence determinants [49]. These plasmids have been associated with the rapid emergence and global spread of blaCTX-M-15, as well as genes encoding resistance to aminoglycosides and fluoroquinolones (e.g. aac(6′)-Ib-cr, qnr, armA, rmtB) [49]. There was an association between blaCTX-M-15 and blaOXA-1, as well as the aac(3)-IIa and aac(6′)-Ib-cr in clade C2 ST131 carrying IncF plasmids, the majority of which came from patients in Harare.
Previous work from North America suggested that the H30-R/C1 clade of ST131 most commonly carries IncF-type F1:A2:B20 plasmids and the H30-Rx/C2 clade is associated with IncF-type F2:A1:B-plasmids [50]. In this study, plasmid types were associated with different sublineages of ST131. For example, IncF-type F36:A4:B1 plasmids were most frequently seen in clade C2, whereas IncF-type F2:A1:B-plasmids were mostly seen in clade C1. A similar observation in ST410 was noted with F1:A1:B49 frequently being observed in B3/H24Rx.
The ST10 CC with blaCTX-M-15 together with other broad-spectrum beta-lactamases, such as blaOXA-1 and blaTEM-1, has been described in clinical isolates from Nigeria [32] and Egypt [51], as well as in poultry from Gambia [52], vegetables from South Africa [39], and enteroaggregative E. coli (EAEC) from Nigeria [53]. EAEC is an established diarrhoeagenic pathotype, transmitted either via consumption of meat products and vegetables or through contact with animals, which has been suggested as a potential source of ESBL bacteria causing diarrhoea and UTIs at the same time. EAEC with ExPEC markers belonging to ST10 has been found to be potential agents of UTIs. Recombination events are important in the evolution of pathogenic EAEC-ST10 UTI linked clones [54].
Several of the other clones identified in this study have been detected elsewhere. ST405 and ST648 are emerging clones associated with carbapenemases, specifically New Delhi metallo-beta-lactamases [55]. ST405 E. coli isolates producing CTX-M-3, CTX-M-14, or CTX-M-15 have been described for using self-transmissible IncF plasmids in acquiring different resistance genes within its clonal lineage [56]. ST354 with CTX-M-15 has been described in humans [57] and stray dogs [58], while to the best of the authors’ knowledge, this is the first report on the presence of blaCTX-M-27 and the insertion prophage integrase M27PP1 in this clone. ST155 has been identified in the current study to harbour blaCTX-M-82. This was also observed in a Gambian study reporting the diversity of E. coli isolates from backyard chickens and guinea fowl; 32% (22/68) of the isolates harboured blaCTX-M-82 [52]. ST354 and ST155 might be strains, which can be exchanged between animals and humans.
This study offers a better understanding of the epidemiology of Zimbabwean ESBL-producing ST131 and ST410 and there close relationship to internationally disseminating ST131 and ST410 strains. It is evident that the Zimbabwe ST131 and ST410 strains are part of the international lineages and that several introductions combined with national transmission have formed the current E. coli population. The clonal nature of the ST131 and ST410 lineage, with highly conserved plasmids in some sublineages, complicates estimation of local circulation and transmission, and highlights the importance of the space time epidemiological link events in the genomic era. However, very close genetic relationships (10–15 SNPs) could indicate a direct transmission even if the epidemiological link is unknown.
Improved strategies for the control of these clones will impact positively on public health management. A number of antimicrobial resistance control strategies have been put in place by different countries. These strategies include (i) improvement of awareness and understanding of antimicrobial resistance through effective communication, education, and training; (ii) strengthening the knowledge and evidence base through surveillance and research; (iii) reduction of the incidence of infection through effective sanitation, hygiene, and infection prevention measures; (iv) optimization of the use of antimicrobial medicines in human and animal health; and (v) the development of the economic case for sustainable investment that takes account of the needs of all countries, and increased investment in new medicines, diagnostic tools, vaccines, and other interventions [59].
The One Health approach is an important initiative for all countries especially in the sub-Saharan countries where information is scarce. The significance of this study from Zimbabwe was to define the scope of the resistance problem. A limitation of this study was the small sample size of only 48 ESBL E. coli isolates with most isolates selected from Harare; however, results from this study contributed to the baseline molecular information for isolates/clones currently and previously linked to resistance in Zimbabwe. Short-read plasmid profile analysis was explored, which does not give a full description of the plasmids as compared to long read sequencing, although this technique provides some indication on the different plasmids within each isolate. Plasmid conjugation experiments could not be performed due to funding and time constraints; therefore, future studies should focus on such experiments to get an in-depth understanding of the characteristics of the ESBL-encoding plasmids. As far as we are aware, this is the first study to be done in Zimbabwe to provide baseline data on virulence, antimicrobial resistance, and detection of specific lineages of ExPEC circulating in our communities using WGS.
Conclusion
Our study has shown a high level of E. coli diversity in terms of STs, antimicrobial resistance genes, serotypes, and virulence genes, which underlines the necessity for concerted efforts for continuous surveillance of the ESBL-producing ExPEC clones. Targeting specific STs (e.g. ST131, ST410, and ST405) through control programs will substantially decrease the spread of ESBLs among ExPEC.
Availability of data and material
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher. All short reads and assemblies associated with this study are available at NCBI under BioProject: PRJNA721804; individual BioSamples are listed in Supplementary File 3.
References
Peirano G, Pitout JDD (2019) Extended-spectrum β-lactamase-producing Enterobacteriaceae: update on molecular epidemiology and treatment options. Drugs 79(14):1529–1541. https://doi.org/10.1007/s40265-019-01180-3
Centers for Disease Control and Prevention (2019) ESBL-producing Enterobacterales in healthcare settings. Centers for Disease Control and Prevention. https://www.cdc.gov/hai/organisms/ESBL.html. Accessed 11 Apr 2019
Pishtiwan AH, Khadija KM (2019) Prevalence of blaTEM, blaSHV, and blaCTX-M genes among ESBL-producing Klebsiella pneumoniae and Escherichia coli isolated from thalassemia patients in Erbil, Iraq. Mediterr J Hematol Infect Dis 11(1):e2019041. https://doi.org/10.4084/MJHID.2019.041
Brolund A (2014) Overview of ESBL-producing Enterobacteriaceae from a Nordic perspective. Infect Ecol Epidemiol 1:4. https://doi.org/10.3402/iee.v4.24555
Mathers AJ, Peirano G, Pitout JD (2015) The role of epidemic resistance plasmids and international high-risk clones in the spread of multidrug-resistant Enterobacteriaceae. Clin Microbiol Rev 28(3):565–591. https://doi.org/10.1128/CMR.00116-14
Pitout JDD, Finn TJ (2020) The evolutionary puzzle of Escherichia coli ST131. Infect Genet Evol 81:104265. https://doi.org/10.1016/j.meegid.2020.104265
Pitout JD, DeVinney R (2017) Escherichia coli ST131: a multidrug-resistant clone primed for global domination. F1000Res 28(6):F1000 Faculty Rev-195. https://doi.org/10.12688/f1000research.10609.1
Birgy A, Bidet P, Levy C, Sobral E, Cohen R, Bonacorsi S (2017) CTX-M-27-Producing Escherichia coli of sequence type 131 and clade C1–M27, France. Emerg Infect Dis 23(5):885. https://doi.org/10.3201/eid2305.161865
Matsumura Y, Pitout JD, Gomi R, Matsuda T, Noguchi T, Yamamoto M, Peirano G, DeVinney R, Bradford PA, Motyl MR, Tanaka M, Nagao M, Takakura S, Ichiyama S (2016) Global Escherichia coli sequence type 131 clade with blaCTX-M-27 Gene. Emerg Infect Dis 22(11):1900–1907. https://doi.org/10.3201/eid2211.160519
Abrar S, Hussain S, Khan RA, Ul Ain N, Haider H, Riaz S (2018) Prevalence of extended-spectrum-β-lactamase producing Enterobacteriaceae: first systematic meta-analysis report from Pakistan. Antimicrob Resist Infect Control 7:26. https://doi.org/10.1186/s13756-018-0309-1
Hu Y, Matsui YW, Riley L (2020) Risk factors for fecal carriage of drug-resistant Escherichia coli: a systematic review and meta-analysis. Antimicrob Resist Infect Control 9(1):31. https://doi.org/10.1186/s13756-020-0691-3
Peirano G, van Greune CH, Pitout JD (2011) Characteristics of infections caused by extended-spectrum β-lactamase-producing Escherichia coli from community hospitals in South Africa. Diagn Microbiol Infect Dis 69(4):449–453. https://doi.org/10.1016/j.diagmicrobio.2010.11.011
Seni J, Peirano G, Okon KO, Jibrin YB, Mohammed A, Mshana SE, DeVinney R, Pitout JD (2018) The population structure of clinical extra-intestinal Escherichia coli in a teaching hospital from Nigeria. Diagn Microbiol Infect Dis 92(1):46–49. https://doi.org/10.1016/j.diagmicrobio.2018.04.001
Sonda T, Kumburu H, van Zwetselaar M, Alifrangis M, Mmbaga BT, Aarestrup FM, Kibiki G, Lund O (2018) Whole genome sequencing reveals high clonal diversity of Escherichia coli isolated from patients in a tertiary care hospital in Moshi, Tanzania. Antimicrob Resist Infect Control 7(1):1–12. https://doi.org/10.1186/s13756-018-0361-x
Estaleva C, Zimba TF, Sekyere JO, Govinden U, Chenia HY, Simonsen GS, Haldorsen B, Essack S, Sundsfjord A (2021) High prevalence of multidrug resistant ESBL- and plasmid mediated AmpC-producing clinical isolates of Escherichia coli at Maputo Central Hospital, Mozambique. BMC Infect Dis 21(16). https://doi.org/10.1186/s12879-020-05696-y
Kabugo D, Kizito S, Ashok DD, Graham KA, Nabimba R, Namunana S, Kabaka MR, Achan B, Najjuka FC (2016) Factors associated with community-acquired urinary tract infections among adults attending assessment centre, Mulago Hospital Uganda. Afr Health Sci 16(4):1131–1142. https://doi.org/10.4314/ahs.v16i4.31
Clinical and Laboratory Standards Institute (2018) Performance standards for antimicrobial susceptibility testing, 28th ed.; CLSI Supplement Document M100; CLSI: Wayne, PA, USA
Technical Manual Wizard® Genomic DNA Purification Kit Wizard® Genomic DNA Purification Kit [internet] (Wisconsin: Promega Corporation). Available from: https://ita.promega.com/media/files/resources/protocols/technical-manuals/0/wizard-genomic-dna-purification-kit-protocols.pdf. [Cited 2017 September 4]
O’Connor AH, Moon TS (2016) Development of design rules for reliable antisense RNA behavior in E. coli. ACS Synth Biol 5(12):1441–1454. https://doi.org/10.1021/acssynbio.6b00036
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19(5):455–477. https://doi.org/10.1089/cmb.2012.0021
Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics 29(8):1072–1075. https://doi.org/10.1093/bioinformatics/btt086
Stamatakis A (2014) RAxML Version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A (2019) RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35(21):4453–4455. https://doi.org/10.1093/bioinformatics/btz305
Rambaut A (2010) FigTree v1.3.1. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 14 Dec 2009
Croucher NJ, Didelot X (2015) The application of genomics to tracing bacterial pathogen transmission. Curr Opin Microbiol 23:62–67. https://doi.org/10.1016/j.mib.2014.11.004
Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M (2006) Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol 60(5):1136–1151. https://doi.org/10.1111/j.1365-2958.2006.05172.x
Beghain J, Bridier-Nahmias A, Le Nagard H, Denamur E, Clermont O (2018) ClermonTyping: an easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb Genom 4(7):e000192. https://doi.org/10.1099/mgen.0.000192
Hunt M, Mather AE, Sánchez-Busó L, Page AJ, Parkhill J, Keane JA, Harris SR (2017) ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microb Genom 3(10):e000131. https://doi.org/10.1099/mgen.0.000131
Liu B, Zheng DD, Jin Q, Chen LH, Yang J (2019) VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res 47(D1):D687–D692
Joensen KG, Tetzschner AMM, Iguchi A, Aarestrup FM, Scheutz F (2015) Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J Clin Microbiol 53:2410–2426. https://doi.org/10.1128/JCM.00008-15
Roer L, Overballe-Petersen S, Hansen F, Schønning K, Wang M, Røder BL, Hansen DS, Justesen US, Andersen LP, Fulgsang-Damgaard D, Hopkins KL, Woodford N, Falgenhauer L, Chakraborty T, Samuelsen Ø, Sjöström K, Johannesen TB, Ng K, Nielsen J, Ethelberg S, Stegger M, Hammerum AM, Hasman H (2018) Escherichia coli sequence type 410 is causing new international high-risk clones. mSphere 3(4):e00337-18. https://doi.org/10.1128/mSphere.00337-18
Sonda T, Kumburu H, van Zwetselaar M, Alifrangis M, Lund O, Kibiki G, Aarestrup FM (2016) Meta-analysis of proportion estimates of extended-spectrum-beta-lactamase-producing Enterobacteriaceae in East Africa hospitals. Antimicrob Resist Infect Control 5:18. https://doi.org/10.1186/s13756-016-0117-4
Storberg V (2014) ESBL-producing Enterobacteriaceae in Africa - a non-systematic literature review of research published 2008–2012. Infect Ecol Epidemiol 13:4. https://doi.org/10.3402/iee.v4.20342
Muvunyi CM, Masaisa F, Bayingana C, Mutesa L, Musemakweri A, Muhirwa G, Claeys GW (2011) Decreased susceptibility to commonly used antimicrobial agents in bacterial pathogens isolated from urinary tract infections in Rwanda: need for new antimicrobial guidelines. Am J Trop Med Hyg 84(6):923–928. https://doi.org/10.4269/ajtmh.2011.11-0057
Muhammad MH, Swedan S (2015) Molecular and phenotypic characterization of carbapenem resistance and extended spectrum beta-lactamases among urinary Escherichia coli isolates. Int J Sci Technol 5(9):1–8
Ogefere HO, Aigbiremwen PA (2015) Omoregie R (2015) Extended spectrum beta-lactamase (ESBL) producing Gram-negative isolates from urine and wound specimens in a tertiary health facility in southern Nigeria. Trop J Pharm Res 14(6):1089–1094
Peirano G, Pitout JD (2010) Molecular epidemiology of Escherichia coli producing CTX-M beta-lactamases: the worldwide emergence of clone ST131 O25:H4. Int J Antimicrob Agents 35(4):316–321. https://doi.org/10.1016/j.ijantimicag.2009.11.003
Albinu I, Odugbemi T, Koenig W, Ghebremedhin B (2012) Sequence type ST131 and ST10 complex (ST617) predominant among CTX-M-15-producing Escherichia coli isolates from Nigeria. Clin Microbiol Infect 18(3):E49-51. https://doi.org/10.1111/j.1469-0691.2011.03730
Ben Slama K, Ben Sallem R, Jouini A, Rachid S, Moussa L, Sáenz Y, Estepa V, Somalo S, Boudabous A, Torres C (2011) Diversity of genetic lineages among CTX-M-15 and CTX-M-14 producing Escherichia coli strains in a Tunisian hospital. Curr Microbiol 62(6):1794–1801. https://doi.org/10.1007/s00284-011-9930-4
Eibach D, Campos CB, Krumkamp R, Al-Emran HM, Dekker D, Boahen KG, Kreuels B, Adu-Sarkodie Y, Aepfelbacher M, Park SE, Panzner U, Marks F, May J (2016) Extended spectrum beta-lactamase producing Enterobacteriaceae causing bloodstream infections in rural Ghana, 2007–2012. Int J Med Microbiol 306:249–254. https://doi.org/10.1016/j.ijmm.2016.05.006
Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ (2015) Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol 13(5):269–284. https://doi.org/10.1038/nrmicro3432
Wilmore SMS, Kranzer K, Williams A, Makamure B, Nhidza AF, Mayini J, Bandason T, Metcalfe J, Nicol MP, Balakrishnan I, Ellington MJ, Woodford N, Hopkins S, McHugh TD, Ferrand RA (2017) Carriage of extended-spectrum beta-lactamase-producing Enterobacteriaceae in HIV-infected children in Zimbabwe. J Med Microbiol 66(5):609–615. https://doi.org/10.1099/jmm.0.000474
Bevan ER, Jones AM, Hawkey PM (2017) Global epidemiology of CTX-M β-lactamases: temporal and geographical shifts in genotype. J Antimicrob Chemother 72(8):2145–2155. https://doi.org/10.1093/jac/dkx146
Berendes D, Kirby A, Brown J, Wester AL (2020) Human faeces-associated extended-spectrum beta-lactamase-producing Escherichia coli discharge into sanitation systems in 2015 and 2030: a global and regional analysis. Lancet Planet Health 4(6):E246–E255. https://doi.org/10.1016/S2542-5196(20)30099-1
Mbelle NM, Feldman C, Osei Sekyere J, Maningi NE, Modipane L, Essack SY (2020) The resistome, mobilome, virulome and phylogenomics of multidrug-resistant Escherichia coli clinical isolates from Pretoria, South Africa. Sci Rep 10:1–16. https://doi.org/10.1038/s41598-020-58160-x
Silva KC, Moreno M, Cabrera C, Spira B, Cerdeira L, Lincopan N, Moreno AM (2016) First characterization of CTX-M-15-producing Escherichia coli strains belonging to sequence type (ST) 410, ST224, and ST1284 from commercial swine in South America. Antimicrob Agents Chemother 60(4):2505–2508. https://doi.org/10.1128/AAC.02788-15
Ben Yahia H, Ben Sallem R, Tayh G, Klibi N, Ben Amor I, Gharsa H, Boudabbous A, Ben SK (2018) Detection of CTX-M-15 harboring Escherichia coli isolated from wild birds in Tunisia. BMC Microbiol 18(1):26. https://doi.org/10.1186/s12866-018-1163-2
da Costa PM, Oliveira M, Bica A, Vaz-Pires P, Bernardo F (2007) Antimicrobial resistance in Enterococcus spp. and Escherichia coli isolated from poultry feed and feed ingredients. Vet Microbiol 120(1–2):122–131. https://doi.org/10.1016/j.vetmic.2006.10.005
Villa L, García-Fernández A, Fortini D, Carattoli A (2010) Replicon sequence typing of IncF plasmids carrying virulence and resistance determinants. J Antimicrob Chemother 65(12):2518–2529. https://doi.org/10.1093/jac/dkq347
Johnson JR, Johnston B, Thuras P, Launer B, Sokurenko EV, Miller LG (2016) Escherichia coli sequence type 131 H30 is the main driver of emerging extended-spectrum-β-lactamase-producing E. coli at a tertiary care center. mSphere 1(6):e00314-16. https://doi.org/10.1128/mSphere.00314-16
Fam N, Leflon-Guibout V, Fouad S, Aboul-Fadl L, Marcon E, Desouky D, El-Defrawy I, Abou-Aitta A, Klena J, Nicolas-Chanoine MH (2011) CTX-M-15-producing Escherichia coli clinical isolates in Cairo (Egypt), including isolates of clonal complex ST10 and clones ST131, ST73, and ST405 in both community and hospital settings. Microb Drug Resist 17(1):67–73. https://doi.org/10.1089/mdr.2010.0063
Foster-Nyarko E, Alikhan NF, Ravi A, Thomson NM, Jarju S, Kwambana-Adams BA, Secka A, O’Grady J, Antonio M, Pallen MJ (2020) Genomic diversity of Escherichia coli isolates from backyard chickens and guinea fowl in the Gambia. Microb Genom 7(1). https://doi.org/10.1099/mgen.0.000484
Okeke IN, Wallace-Gadsden F, Simons HR, Matthews N, Labar AS, Hwang J, Wain J (2010) Multi-locus sequence typing of enteroaggregative Escherichia coli isolates from Nigerian children uncovers multiple lineages. PLoS ONE 5(11):e14093. https://doi.org/10.1371/journal.pone.0014093
Chattaway MA, Jenkins C, Ciesielczuk H, Day M, DoNascimento V, Day M, Rodríguez I, van Essen-Zandbergen A, Schink AK, Wu G, Threlfall J, Woodward MJ, Coldham N, Kadlec K, Schwarz S, Dierikx C, Guerra B, Helmuth R, Mevius D, Woodford N, Wain J (2014) Evidence of evolving extra-intestinal enteroaggregative Escherichia coli ST38 clone. Emerg Infect Dis 20(11):1935–1937. https://doi.org/10.3201/eid2011.131845
Alonso CA, Zarazaga M, Ben Sallem R, Jouini A, Ben Slama K, Torres C (2017) Antibiotic resistance in Escherichia coli in husbandry animals: the African perspective. Lett Appl Microbiol 64(5):318–334. https://doi.org/10.1111/lam.12724
Hrabák J, Empel J, Bergerová T, Fajfrlík K, Urbásková P, Kern-Zdanowicz I, Hryniewicz W, Gniadkowski M (2009) International clones of Klebsiella pneumoniae and Escherichia coli with extended-spectrum beta-lactamases in a Czech hospital. J Clin Microbiol 47(10):3353–3357. https://doi.org/10.1128/JCM.00901-09
Carattoli A, Bertini A, Villa L, Falbo V, Hopkins KL, Threlfall EJ (2005) Identification of plasmids by PCR-based replicon typing. J Microbiol Methods 63(3):219–228. https://doi.org/10.1016/j.mimet.2005.03.018
Corvec S, Crémet L, Leprince C, Dauvergne S, Reynaud A, Lepelletier D, Caroff N (2010) Epidemiology of Escherichia coli clinical isolates producing AmpC plasmidic beta-lactamase during a 5-year period in a French teaching Hospital. Diagn Microbiol Infect Dis 67(3):277–281. https://doi.org/10.1016/j.diagmicrobio.2010.02.007
World Health Organization (2015) Global action plan on antimicrobial resistance. World Health Organization. https://apps.who.int/iris/handle/10665/193736. Accessed 10 Nov 2015
Acknowledgements
The authors would like to thank the Department of Medical Microbiology, University of Pretoria, SA, and the staff at 14 laboratories from Zimbabwe (Harare Central Hospital, Beatrice Road Infectious Diseases, Parirenyatwa Group of Hospitals, National Microbiology Reference Laboratory, Marondera Central Hospital, Bindura Hospital, Mpilo Hospital, Premier Medical Society Laboratory, CIMAS laboratories, Lancet Laboratories) for their contributions in making the national surveillance a success and contributing to the biobanking of these isolates for future studies. Also, we would like to thank the sequencing team at Quadram Institute Biosciences, UK, for the help and assistance offered.
Funding
This project was funded by the National Health Laboratory Service (NHLS), the University of Pretoria, South Africa, and a strategic partnership between National Microbiology Reference Laboratory and Quadrum Institute Biosciences.
Author information
Authors and Affiliations
Contributions
All authors provided critical input and contributed to the manuscript writing and approved its final version. Conception or design of the work: Faustinos Tatenda Takawira, Johann Pitout, Marleen M. Kock, Sekesai Zinyowera-Mtapurwa, Tapfumanei Mashe, Leckson Mukavhi, Marthie M. Ehlers. Methodology: Faustinos Tatenda Takawira, Johann Pitout, Marleen M. Kock, Jorge Matheu, Stanley Munyaradzi Midzi, Lusubilo Witson Mwamakamba, Andrew Tarupiwa; formal analysis and interpretation: Faustinos Tatenda Takawira, Johann Pitout, Marleen M. Kock, Rob Kingsley, Gaetan Thilliez, Ana Victoria Gutierrez, Gisele Peirano, David Gally, Tapfumanei Mashe; writing—original draft preparation: Faustinos Tatenda Takawira, Johann Pitout, Marleen M. Kock; writing—review and editing of the article: Faustinos Tatenda Takawira, Johann Pitout, Marleen M. Kock, Marthie M. Ehlers, Andrew Tarupiwa, Tapfumanei Mashe; final approval of the version to be published: Faustinos Tatenda Takawira, Johann Pitout, Marleen M. Kock, Sekesai Zinyowera-Mtapurwa, Tapfumanei Mashe, Lusubilo Witson Mwamakamba, Gaetan Thilliez, Ana Victoria Gutierrez, Rob Kingsley, Gisele Peirano, Jorge Matheu, Stanley Munyaradzi Midzi, Leckson Mukavhi, David Gally, Andrew Tarupiwa, Marthie M. Ehlers.
Corresponding author
Ethics declarations
Consent for publication
All authors have read the manuscript and approved submission.
Conflict of interest
The authors declare no competing interests.
Disclaimer
Opinions expressed and conclusions arrived at are those of the authors and are not necessarily to be attributed to the funders. The funders of the study had no role in the study design, data collection, data analysis, interpretation, or writing of the article.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Takawira, F.T., Pitout, J., Thilliez, G. et al. Molecular epidemiology of extended-spectrum beta-lactamase–producing extra-intestinal pathogenic Escherichia coli strains over a 2-year period (2017–2019) from Zimbabwe. Eur J Clin Microbiol Infect Dis (2021). https://doi.org/10.1007/s10096-021-04379-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10096-021-04379-z