
Abstract
The diagnosis of Clostridium difficile infection (CDI) requires the detection of toxigenic C. difficile or its toxins and a clinical assessment. We evaluated the performance of four nucleic acid amplification tests (NAATs) detecting toxigenic C. difficile directly from faeces compared to routine toxigenic culture. In total, 300 faecal samples from Danish hospitalised patients with diarrhoea were included consecutively. Culture was performed in duplicate (routine and ‘expanded toxigenic culture’: prolonged and/or re-culture) and genotypic toxin profiling by polymerase chain reaction (PCR), PCR ribotyping and toxinotyping (TT) were performed on culture-positive samples. In parallel, the samples were analysed by four NAATs; two targeting tcdA or tcdB (illumigene® C. difficile and PCRFast® C. difficile A/B) and two multi-target real-time (RT) PCR assays also targeting cdt and tcdC alleles characteristic of epidemic and potentially more virulent PCR ribotypes 027, 066 and 078 (GeneXpert® C. difficile/Epi and an ‘in-house RT PCR’ two-step algorithm). The multi-target assays were significantly more sensitive compared to routine toxigenic culture (p < 0.05) and significantly more robust to inhibition compared to PCRFast (p < 0.001). Duplicate ‘expanded toxigenic culture’ increased the culture-positive rate by 29 % compared to routine culture. The ability of the GeneXpert and in-house assays to correctly classify PCR ribotype 027 was high (>95 %), and in-house PCR displayed 100 % correct identification of PCR ribotypes 066 and 078. Furthermore, the presence of the PCR enhancer bovine serum albumin (BSA) was found to be related to high sensitivity and low inhibition rate. Rapid laboratory diagnosis of toxigenic C. difficile by RT PCR was accurate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past decade, the prevalence and severity of Clostridium difficile infection (CDI) has increased, due to outbreaks of epidemic strains, notably BI/NAP1/027 [polymerase chain reaction (PCR) ribotype 027] and PCR ribotype 078 [1–3]. Poor patient outcome, treatment failure, and increased mortality and recurrence rates have been associated with PCR ribotypes 027 and 078 [4–6].
The main causative agents of CDI are two well-known large clostridial toxins, toxin A (TcdA) and toxin B (TcdB), expressed from their respective genes, tcdA and tcdB, in the pathogenicity locus (PaLoc) [7]. Regulators of tcdA and tcdB reside within the PaLoc, including a putative negative regulator tcdC [7]. The genotypic characteristics of highly virulent strains of C. difficile are several mutations in the tcdC gene. PCR ribotype 027 harbours an 18-bp in-frame deletion and a single nucleotide (nt) deletion at position 117 (Δ117) in tcdC, whereas PCR ribotype 078 (and 066) possesses a 39-bp in-frame deletion and a single nt substitution at position 184 (C184T) in tcdC [6, 8]. However, the importance of these genotypic changes in tcdC for virulence remains to be determined [9–12]. In addition to tcdA and tcdB, epidemic strains (including PCR ribotypes 027, 066 and 078) may express a third toxin, the binary toxin (actin-ADP-ribosylating toxin C. difficile transferase, CDT), encoded by cdtA and cdtB located in the Cdt locus [13]. The binary toxin genes have been suggested as an additional cause of excess morbidity, mortality and higher recurrence rates of CDI [14, 15].
The presence of tcdA, tcdB and cdtA/cdtB in combination with specific nt mutations in tcdC, expressed by epidemic strains of C. difficile, can be exploited in the laboratory diagnosis of C. difficile. Rapid and reliable diagnosis of CDI is essential for optimal patient management, infection control and understanding the epidemiology. Several fast commercial nucleic acid amplification tests (NAATs) are now available, including seven U.S. Food and Drug Administration (FDA)-approved tests (http://www.fda.gov/medicaldevices/productsandmedicalprocedures/invitrodiagnostics/ucm330711.htm). Conventional PCR, real-time (RT) PCR and loop-mediated isothermal amplification (LAMP) are established NAATs used in laboratory diagnostics of microorganisms. A limitation to PCR detection, when applied to biological samples, is the presence of inhibitors [16], which can result in indeterminate or false-negative results. However, reagents such as bovine serum albumin (BSA) are capable of buffering inhibitors in biological samples [17]. Therefore, BSA has the potential to optimise RT PCR-based diagnostic methods.
The objective of this study was to evaluate the performance of three commercial NAATs, illumigene® C. difficile, GeneXpert® C. difficile/Epi PCR and PCRFast® C. difficile A/B, and an in-house RT PCR algorithm with comparison to our current method, toxigenic culture.
Materials and methods
Study population and sample collection
A total of 300 consecutive faecal samples from 283 hospitalised patients with diarrhoea were enrolled from February 14th to April 5th 2011. Samples were included upon submission for routine diagnostics of C. difficile at the Department of Clinical Microbiology, Slagelse Hospital, Denmark. The criterion for testing was infectious diarrhoea. Multiple samples from the same patient were allowed, with a minimum interval of 14 days between sampling. The samples were sent at ambient temperature without a transport medium. The age inclusion criterion was ≥2 years. However, children <2 years were included if C. difficile testing was specifically requested by the physician.
Toxigenic culture and genotyping
On arrival in the laboratory, the faecal samples were kept at 5 °C until cultures were performed, usually within 4 to 5 h of receipt. The majority (>90 %) of samples were tested within 24 h of collection. For spore purification, 1 ml of stool was suspended in (i) 1 ml of 99.9 % ethanol and incubated for 1 h at ambient temperature or (ii) 1 ml of saline, heated at 80 °C for 10 min and cooled for 5 min at ambient temperature. Approximately 50–75 μl of stool suspension was plated on cycloserine cefoxitin fructose agar (CCFA) plates (SSI Diagnostica, Hillerød, Denmark) and incubated under anaerobic conditions at 37 °C for 48 h. Anaerobic culture was performed in duplicate; one plate for the routine and one for the research diagnostics. Presumptive C. difficile colonies were identified by standard phenotypic characteristics: yellow colour; flat, filamentous, snowflake-like colony morphology; ‘horse-barn’ odour (paracresol); and, when indeterminate, positive L-proline aminopeptidase test (Rosco Diagnostica, Taastrup, Denmark). Routine diagnostic results were blinded to the NAATs testing. All negative research plates at 48 h were re-incubated for an additional 7–10 days. Continued culture-negative but NAAT-positive samples were re-cultured with an extended incubation time (7–10 days). Prolonged and/or re-culture results are represented by the ‘expanded culture’ reference method.
Culture-positive samples were analysed for their toxigenic nature by toxin profiling, PCR ribotyping and toxinotyping (TT). These tests were performed blinded to the NAATs results. One to five (I–V) C. difficile colonies from the research plate were sub-cultured individually onto 5 % horse blood agar plates (SSI Diagnostica) and incubated anaerobically at 37 °C for 24–72 h. Isolates were stored in beef broth with 10 % glycerol (SSI Diagnostica) at −80 °C. Following the inclusion period, one isolate per episode was recovered and genotypic toxin profiling of tcdA, tcdB and cdtA/cdtB was performed as previously published [18, 19]. In addition, the in-frame deletions of tcdC were detected according to Persson et al. [19]. PCR ribotyping was carried out according to O’Neill et al. [20] and Stubbs et al. [21]. Unknown PCR ribotypes (no profile match in the reference strain collection) were designated SLAxxxx. TT, based on the restriction patterns of tcdB and tcdA using restriction fragment length polymorphism PCR, was done according to Rupnik et al. [22]. When the obtained genotypic results from GeneXpert and/or the in-house PCR diverged in comparison to the genotypic toxin profiling and PCR ribotyping, additional isolates from the specific sample were analysed, if available.
NAATs
Two automated FDA-approved tests with integrated DNA extraction (provided in the kit) and nucleic acid amplification (NAA) protocols and two manual tests with independent DNA extraction and PCR protocols were evaluated. The automated tests were illumigene (Meridian Bioscience, Milan, Italy) and GeneXpert (Cepheid, Sunnyvale, CA, USA); the manual tests were PCRFast (ifp, Institut für Produktqualität, GmbH, Berlin, Germany) and one in-house RT PCR two-step algorithm [23, 24]. Toxigenic culture, anaerobic culture followed by genotypic toxin profiling combined with PCR ribotyping, was used as the reference method to evaluate the performance of the four NAATs.
illumigene and PCRFast detect tcdA and tcdB only and, thus, provide a toxigenic C. difficile-positive or -negative answer. According to the absence or presence of cdt and Δ117tcdC, GeneXpert can differentiate toxigenic C. difficile-positives in non-027 or presumptive PCR ribotype 027. The in-house PCR was designed to classify toxigenic C. difficile-positive samples in non-027, presumptive PCR ribotype 027 or potential 066 or 078 (066/078) on the basis of cdtA +/− and specific single nt mutations in the tcdC gene.
Automated NAATs and faecal suspension preparation for independent DNA extraction were performed daily, and that for the manual NAATs in batches weekly.
For the three commercial tests, execution, results interpretation and resolving of indeterminate results (including inhibited samples) were performed according to the manufacturers’ guidance and by the visual inspection of raw data (amplification curves, Ct values and level of fluorescence).
Automated NAATs
illumigene is based on LAMP, whereas GeneXpert is a multiplex RT PCR assay. For illumigene, external quality control was performed once a day and for GeneXpert for each lot of kits using a negative control (NC) (w/o stool) and a positive control (PC) (toxigenic C. difficile faecal sample). Inhibited samples were repeated with a reduced faecal load until a valid result was recorded.
Independent DNA extraction for manual NAATs: NucliSENS® easyMAG®
Nucleic acid extraction from stool was performed on the NucliSENS® easyMAG® platform (bioMérieux, Marcy I’Etoile, France), according to the manufacturer’s protocol optimised for stool samples (“Extraction Protocol for the NucliSENS easyMAG BTL039444 rel. 1.0 for stool samples” in combination with “Specific B” protocol). Stool samples were transferred to NucliSENS® Lysis Buffer (bioMérieux) [1:2 (wt/vol)], vortexed and homogenised for 1 min at 7,000 rpm using the MagNA Lyser Instrument (Roche Applied Science, Penzberg, Germany). Faecal suspensions were centrifuged for 2 min at 16,000 g and stored at −20 °C until batch testing. The automated extraction was conducted in batches of 24 samples. 100 μl of thawed and re-spun supernatant was incubated in 2 ml of NucliSENS® Lysis Buffer for 10 min at room temperature, including phocine herpesvirus (PhHV) [25] serving as the internal extraction and amplification control (IC) in the in-house algorithm. This total volume of 2.1 ml together with 140 μl of magnetic silica was loaded onto the platform. The output eluates (110 μl) were stored at 5 °C until weekly manual RT PCR analysis.
Manual NAAT: PCRFast® C. difficile
PCRFast is an RT PCR test amplifying tcdA, tcdB and an IC. All input eluates were initially diluted 1:4 in nuclease-free water (Qiagen) in order to prevent total inhibition (this is in accordance with the protocol). NC (water) and PCs (supplied in the kit) were included in each batch. Inhibited samples (no signal from the IC) were resolved by further dilution of the DNA input, until a valid result (signal from the IC) was obtained.
Manual NAAT: in-house multiplex RT PCR two-step algorithm
The in-house algorithm consists of two RT PCR steps; a ‘toxin’ followed by a ‘tcdC genotypic’ reaction. The algorithm was adapted from previously published primer and probe designs [23, 24], with optimisations and modifications compatible with our facility, including the introduction of new probes.
The toxin reaction detects tcdA, tcdB and cdtA, and serves as a screening assay [23]. The toxin reaction was performed on all 300 faecal samples, whereas tcdC genotyping for three tcdC alleles [wild type (wt), Δ117 deletion and an A117T point mutation] was carried out on toxin gene-positive samples only, although including NAAT-negative but culture-positive samples.
In our hands, the original probe designs by de Boer et al. [24] and Hoegh et al. [23] produced low fluorescence and linear amplification curves in pilot testing (data not shown). However, for the toxin reaction, the addition of a master mix optimised for multiplex RT PCR in combination with BSA produced conclusive results.
For the tcdC genotyping reaction, new probes were designed to improve specificity and enable the identification of presumptive C. difficile PCR ribotypes 066 and 078. Sequence analysis of tcdC gene sequences available from GenBank identified an A117T mutation in tcdC, which results in a truncated open reading frame. In previously tcdC sequenced strains, we found this variant in epidemic PCR ribotypes as 066 and 078, although not exclusively. The binary toxin-positive PCR ribotype 023 was seen to carry this mutation too. The new probe set makes it possible to distinguish the A117T signal from the Δ117 signal. Both mutation probes will generate a signal in the presence of either the Δ117 deletion or the A117T point mutation in tcdC; however, the ‘correct’ probe will yield the strongest signal (lowest Ct value), enabling discrimination. Usually, wt samples will yield a single tcdC signal exclusively. Thus, the tcdC genotyping, in combination with the toxin reaction, separates C. difficile non-027, presumptive 027 and potential 066/078 PCR ribotypes. The primer and probe sequences and final concentrations are given in Table 1. PCR reactions contained 5 μl of eluate in a total volume of 20 μl (toxin reaction) or 25 μl (tcdC genotyping) and 0.2 μg/μl BSA (Fermentas, Ontario, Canada). The toxin reaction contained the 2× TaqMan Fast Advanced Master Mix (Applied Biosystems, Foster City, CA, USA), whereas the tcdC genotyping reaction contained the originally reported 2× TaqMan Universal PCR Master Mix (Applied Biosystems) [24]. DNA extracts from a negative (NC), a non-027 and a PCR ribotype 027-positive C. difficile faecal sample (PCs) served as controls in each batch test. PCR was performed on an Mx3005P thermocycler (Stratagene, La Jolla, CA, USA) using the following conditions: 2 min at 50 °C, 10 min at 95 °C; followed by 42/40 cycles of 15 s at 95 °C and 1 min at 60 °C. Batch RT PCR was considered valid when the NC generated the IC signal and the two PCs generated signals according to their genotypes. Patient specimens were considered positive when two or more signals, toxin and tcdC genotyping combined, reached a fixed threshold of 0.05. An RT PCR was considered inhibited when no signal from the IC was recorded.
Statistics
For the statistical comparison of routine toxigenic culture-negative samples, a regression method for paired samples was used [26]. The outputs were relative positive fractions with 95 % confidence intervals (CIs). For tabulated data with low or zero counts, Fisher’s exact test and McNemar’s test were used. A p-value of 0.05 was considered significant. The statistical calculations were performed in the open access software R. The Fisher test command in R allows for tables larger than 2 × 2.
Ethical approval
This study obtained approval from the Regional Ethics Committee of Zealand, Denmark (SJ-208). Informed patient consent was waived.
Results
Routine toxigenic culture as the reference method
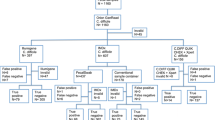
A total of 300 consecutive faecal samples from 283 patients were tested for C. difficile by duplicate anaerobic culture and four NAATs. One patient contributed three samples and 15 patients contributed two samples. Two patients under the age of 2 years were included. The median age of the 161 female and 122 male patients was 66 years (range 4 months to 97 years).
In routine testing, anaerobic culture yielded 42 C. difficile-positive samples from 39 patients (median age 69.8 years; range 6–95 years). C. difficile isolated from three samples were non-toxigenic by genotypic PCR, and equally found negative by the four NAATs and were classified as negative. One isolate (F-5830) could not be recovered and was excluded from analysis. Hence, the prevalence of toxigenic C. difficile estimated by routine culture was 12.7 % (38 out of 299).
Forty-nine faecal specimens yielded an initially invalid (inhibited) test result in one or two NAAT assays: none for in-house PCR, two with GeneXpert, eight with illumigene and 40 with PCRFast (Table 2). One specimen (F-4350) gave an invalid result in both illumigene and PCRFast, whereas the other 48 invalid results were recorded in one assay only. The majority (98.0 %) of invalid results were recorded from C. difficile culture-negative samples. The total inhibition rate of PCRFast (13.4 %) was significantly (p < 0.001) higher than the rate of inhibition produced by the other assays (Table 2). Specimens yielding an initially invalid result were re-tested with diluted template or reduced faecal load until a valid result was recorded, and this result was used in performance characterisation.
Using routine toxigenic culture as the reference method for comparison, the sensitivity and specificity of the four NAATs are given in Table 3. The overall agreement (accuracy) with routine toxigenic culture was similar for all four NAATs; 92.6 %, 93.3 %, 95.3 % and 95.7 % for the in-house PCR, GeneXpert, PCRFast and illumigene assay, respectively (Table 3). The specificity ranged from 92.0 % to 98.1 %, with no significant difference between tests. The highest specificity was recorded for the PCRFast assay. However, this assay had a significantly (p < 0.001) lower sensitivity (76.3 %). illumigene, in-house PCR and GeneXpert had sensitivities of 94.7 %, 97.4 % and 100 %, respectively. The false-negative samples (routine toxigenic culture-positive samples that failed to be detected by an NAAT) are given in Table 4. One sample failed to be detected in three NAATs, one sample failed in two NAATs and seven samples failed in PCRFast only.
As presented in Table 3, it is evident that routine toxigenic culture missed some C. difficile-positive samples. Three of the four NAATs identified more C. difficile-positive samples than routine toxigenic culture; illumigene, GeneXpert and the in-house PCR. The two multi-target assays were significantly more sensitive than routine toxigenic culture. Overall, for all 299 samples, 38 (12.7 %) were positive in routine toxigenic culture. The GeneXpert and in-house PCR gave 58 (19.4 %) positive results, significantly increasing the diagnostic yield with a relative positive fraction of 1.53 (95 % CI 1.05–2.24, p = 0.03). Using the positive fraction of expanded toxigenic culture as the reference, no significant difference in specificity was found (Table 3).
‘Expanded toxigenic culture’ as the reference method
Besides identifying all routine positive samples as C. difficile-positives, expanded culture (duplicate plating, prolonged incubation time and re-culture of NAAT-positive but routine culture-negative samples) resolved an additional 12 samples as C. difficile culture-positives (Table 5). Two strains were excluded; one was non-cultivable after storage (F-3618) and one was non-toxigenic (F-5508) by genotypic PCR, giving a prevalence of 16.1 % when the expanded culture is included (48 out of 299). Seven samples remained culture-negative but NAAT-positive by at least two NAATs. In all, 16 routine culture-negative samples were resolved as true-positives (TPs) by the use of a composite reference (positive in at least two NAATs) (Table 5).
Seven of the ten additional toxigenic culture-positive strains belonged to PCR ribotype 027, two were other PCR ribotypes (SLA0014 and 078) and one sample (F-5957) was polymicrobial, containing two PCR ribotypes, 014/020/077 and SLA0011 (non-toxigenic strain) (Table 5). GeneXpert detected all additional samples as positives. In-house PCR detected all but the polymicrobial sample. illumigene and PCRFast detected only 50 % and 20 % of the additional samples, respectively.
Screening for presumptive 027 and potential 066/078 PCR ribotypes
To assign the GeneXpert and in-house PCR assays’ ability to identify presumptive 027 and 066/078 PCR ribotypes, the typing results from the expanded culture was used as the reference method (Supplemental Table S1). Two specimens (F-5568 and F-5957) were polymicrobial and, therefore, excluded in this context. Fifty-four percent of the total of 46 toxigenic strains belonged to PCR ribotype 027; 17.4 % were 014/020/077 and the remaining were various other PCR ribotypes, including ten previously unknown. The study included a total of two 066 and 078 PCR ribotypes. The GeneXpert and in-house PCR assays identified all 027 faecal samples as positive for toxigenic C. difficile, although the in-house PCR incorrectly assigned one sample as non-027. PCRFast classified 40.0 % (10) of 027 samples as negative. All non-027-066-078 PCR ribotype samples were correctly identified by the GeneXpert assay. One, three and six of the non-027-066-078 PCR ribotype samples failed to be detected by the in-house PCR, illumigene and PCRFast assays, respectively. These samples represented six different PCR ribotypes (014/020/077, 095, SLA0005, SLA0008, SLA0010, SLA0014) belonging to toxinotype 0 (Supplemental Table S1). The in-house PCR design’s ability to recognise a mutation in tcdC characteristic of presumptive 066 and 078 PCR ribotypes was 100 % correct (n = 2). illumigene, GeneXpert and PCRFast identified the 066 and 078 samples as positive; however, GeneXpert incorrectly classified the 078 sample as presumptive 027. The overall agreement between GeneXpert and in-house PCR and PCR ribotyping was high (>95 %). No correlation between discordant results and strain type was found for any of the four NAATs.
Discussion
The optimal laboratory diagnosis of CDI remains an area of controversy [27–29]. In addition, the best suited gold standard method for comparative studies remains to be determined. If the detection of toxigenic strains is the aim, then culture with demonstration of the toxigenic potential of the isolate is the appropriate reference method [27]. The diagnosis of CDI requires both clinical assessment and the laboratory detection of toxigenic C. difficile. Thus, laboratory testing should be limited to patients with clinical symptoms compatible with CDI, and the goal of the laboratory is to quickly and accurately ascertain whether toxigenic C. difficile is present [27].
Three of the four evaluated NAATs identified more positive samples than routine toxigenic culture; illumigene, GeneXpert and in-house PCR. The two multi-target assays were significantly more sensitive than toxigenic culture. The low sensitivity of our toxigenic culture was surprising, but may be explained by aerobic toxicity, non-cultivable organisms in the sample, our routine spore enrichment method and culture medium (CCFA), which has been reported to be sub-optimal [30]. Direct plating of stool samples on a chromogenic agar has been reported to be potentially more sensitive than growth on cefoxitin cycloserine media for C. difficile [31, 32]. However, since samples could be resolved by expanded toxigenic culture and high agreement between NAATs was observed, the data suggested that the initial culture-negative but NAAT-positive samples were, indeed, TPs. This is in agreement with the observation that broth enrichment and duplicate culturing may enhance the C. difficile culture-positive rate by 30 % [33]. The clinical significance of a positive NAAT as a stand-alone method has been questioned; recently, however, RT PCR detection of toxigenic C. difficile by GeneXpert has been reported to correlate with the clinical diagnosis of CDI [29].
Conversely, the inability of an NAAT to detect a culture-positive sample could be a result of poor assay test design, inhibition, limit of detection etc. Furthermore, in the present study, two polymicrobial (multiple strains) samples were found, both yielding inconsistent results between NAATs. Multiple PCR ribotypes of C. difficile may be present simultaneously [34, 35], and this might explain deviations between PCR ribotyping and NAAT results, and between different NAATs.
Compared to enzyme immunoassay (EIA) for glutamate dehydrogenase (GDH), GeneXpert has been reported to have a higher sensitivity for non-027 versus 027 PCR ribotype samples [36], and EIAs to have a higher sensitivity for 027 versus non-027 [36, 37]. However, on cultured isolates, GDH and GeneXpert have been reported not to be affected by specific PCR ribotypes [38]. In accordance, our data showed no difference in the identification of PCR ribotypes, emphasising the importance of a mutual reference method.
The objective of the present study was to evaluate the diagnostic performance on consecutive clinical samples of four NAATs addressing toxin targets compared to toxigenic culture of C. difficile. Two of the assays could provide additional information regarding the presence of cdt and specific single nt mutations in tcdC (Δ117 and A117T) characteristic of potential epidemic strains, such as 027, 066 and 078 PCR ribotypes, and their performance in recognising potential 027 PCR ribotype was high. For GeneXpert, this is in agreement with studies from the United States [39, 40]. In Europe, PCR ribotype 027 is epidemic and 078 is the third most common cause of CDI [41, 42]. Further, the in-house algorithm displayed correct identification of PCR ribotypes 066 and 078. However, the sample size was too small to be conclusive on these PCR ribotypes specifically; hence, further studies on C. difficile strains with the A117T tcdC allele are warranted. Single nt mutations in tcdC (A117T and C184T) have been shown to predict recurrent CDI [12]. Thus, it would be of interest to include clinical data in a study on the A117T tcdC allele and its significance to CDI.
In this study, superior C. difficile recognition, sensitivity and low inhibition correlated with the presence of the PCR enhancer BSA in the RT PCR reaction. The GeneXpert assay had 0.7 % inhibited samples and the in-house PCR had none. Conversely, PCRFast, not including BSA, displayed a significantly higher rate of inhibition (13.4 %) and a significantly lower sensitivity (76.3 %). Since the in-house PCR utilises the same DNA extract as PCRFast, the high rate of invalid results observed for PCRFast seems to reflect susceptibility to the presence of inhibitors in the template. The protocol for DNA extraction on the easyMAG® platform we used is particularly optimised for stool specimens. In combination with the addition of BSA, the “Specific A” protocol exhibits high efficiency in RT PCR [43]. The “Specific B” protocol used in the present study resembles Specific A, but with six additional washing steps - a further improvement. However, the high inhibition rate of PCRFast indicates that the DNA extraction procedure alone does not manage inhibitors. Faeces are known to contain components that inhibit PCR, e.g. dietary components [44] and heme [45]. BSA and other PCR enhancers have been shown to buffer these [46], an observation which our results seem to support. The PCRFast protocol recommends resolving inhibited samples by further dilution of input DNA, which, again, was reflected in the significantly lower sensitivity. In comparison, the in-house PCR assay managed twice as much DNA as PCRFast without inhibition.
The lower inhibition rate of illumigene compared to that of PCRFast supports the general finding that the LAMP technique is more tolerant to inhibitors in biological samples compared to PCR [47]. In this study however, the BSA-optimised RT PCR assays exhibited an even lower inhibition rate than the illumigene LAMP-based assay. Indeed, recent findings suggest that the susceptibility of different NAATs to inhibitors depends on the particular inhibitor [48]. To our knowledge, illumigene does not contain BSA. Therefore, we speculate that the addition of an inhibition buffer to the illumigene and PCRFast assays (if possible) might increase the robustness of these assays in the presence of inhibitors and increase sensitivity.
In conclusion, the two best performing assays were a commercial kit-based and fully automated system (GeneXpert) and an in-house RT PCR two-step algorithm, which can be used in different laboratory settings. They offer prompt and proper detection of toxigenic C. difficile. Furthermore, they have the ability to differentiate CDI according to the presumptive PCR ribotype. These assays were more sensitive than toxigenic culture, included BSA and were more robust to inhibition compared to the other two NAATs evaluated. With the large number of C. difficile NAATs now commercially available, the presence of a PCR enhancer such as BSA may be an additional factor to consider when selecting a reliable laboratory diagnostic test, besides turn-around-time, costs, hands-on time, laboratory facilities, technical skills etc.
References
Kuijper EJ, Barbut F, Brazier JS, Kleinkauf N, Eckmanns T, Lambert ML et al (2008) Update of Clostridium difficile infection due to PCR ribotype 027 in Europe, 2008. Euro Surveill 13(31)). pii: 18942
Janezic S, Ocepek M, Zidaric V, Rupnik M (2012) Clostridium difficile genotypes other than ribotype 078 that are prevalent among human, animal and environmental isolates. BMC Microbiol 12:48
McDonald LC, Killgore GE, Thompson A, Owens RC Jr, Kazakova SV, Sambol SP et al (2005) An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med 353(23):2433–2441
Walker AS, Eyre DW, Wyllie DH, Dingle KE, Griffiths D, Shine B et al (2013) Relationship between bacterial strain type, host biomarkers, and mortality in Clostridium difficile infection. Clin Infect Dis 56(11):1589–1600
Pépin J, Valiquette L, Cossette B (2005) Mortality attributable to nosocomial Clostridium difficile-associated disease during an epidemic caused by a hypervirulent strain in Quebec. CMAJ 173(9):1037–1042
Goorhuis A, Bakker D, Corver J, Debast SB, Harmanus C, Notermans DW et al (2008) Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin Infect Dis 47(9):1162–1170
Voth DE, Ballard JD (2005) Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18(2):247–263
Curry SR, Marsh JW, Muto CA, O’Leary MM, Pasculle AW, Harrison LH (2007) tcdC genotypes associated with severe TcdC truncation in an epidemic clone and other strains of Clostridium difficile. J Clin Microbiol 45(1):215–221
Murray R, Boyd D, Levett PN, Mulvey MR, Alfa MJ (2009) Truncation in the tcdC region of the Clostridium difficile PathLoc of clinical isolates does not predict increased biological activity of Toxin B or Toxin A. BMC Infect Dis 9:103
Goldenberg SD, French GL (2011) Lack of association of tcdC type and binary toxin status with disease severity and outcome in toxigenic Clostridium difficile. J Infect 62(5):355–362
Verdoorn BP, Orenstein R, Rosenblatt JE, Sloan LM, Schleck CD, Harmsen WS et al (2010) High prevalence of tcdC deletion-carrying Clostridium difficile and lack of association with disease severity. Diagn Microbiol Infect Dis 66(1):24–28
Stewart DB, Berg AS, Hegarty JP (2014) Single nucleotide polymorphisms of the tcdC gene and presence of the binary toxin gene predict recurrent episodes of Clostridium difficile infection. Ann Surg 260(2):299–304
Bouvet PJ, Popoff MR (2008) Genetic relatedness of Clostridium difficile isolates from various origins determined by triple-locus sequence analysis based on toxin regulatory genes tcdC, tcdR, and cdtR. J Clin Microbiol 46(11):3703–3713
Stewart DB, Berg A, Hegarty J (2013) Predicting recurrence of C. difficile colitis using bacterial virulence factors: binary toxin is the key. J Gastrointest Surg 17(1):118–124
Bacci S, Mølbak K, Kjeldsen MK, Olsen KE (2011) Binary toxin and death after Clostridium difficile infection. Emerg Infect Dis 17(6):976–982
Wilson IG (1997) Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol 63(10):3741–3751
Kreader CA (1996) Relief of amplification inhibition in PCR with bovine serum albumin or T4 gene 32 protein. Appl Environ Microbiol 62(3):1102–1106
Persson S, Torpdahl M, Olsen KE (2008) New multiplex PCR method for the detection of Clostridium difficile toxin A (tcdA) and toxin B (tcdB) and the binary toxin (cdtA/cdtB) genes applied to a Danish strain collection. Clin Microbiol Infect 14(11):1057–1064
Persson S, Jensen JN, Olsen KE (2011) Multiplex PCR method for detection of Clostridium difficile tcdA, tcdB, cdtA, and cdtB and internal in-frame deletion of tcdC. J Clin Microbiol 49(12):4299–4300
O’Neill GL, Ogunsola FT, Brazier JS, Duerden BI (1996) Modification of a PCR ribotyping method for application as a routine typing scheme for Clostridium difficile. Anaerobe 2(4):205–209
Stubbs SL, Brazier JS, O’Neill GL, Duerden BI (1999) PCR targeted to the 16S-23S rRNA gene intergenic spacer region of Clostridium difficile and construction of a library consisting of 116 different PCR ribotypes. J Clin Microbiol 37(2):461–463
Rupnik M, Avesani V, Janc M, von Eichel-Streiber C, Delmée M (1998) A novel toxinotyping scheme and correlation of toxinotypes with serogroups of Clostridium difficile isolates. J Clin Microbiol 36(8):2240–2247
Hoegh AM, Nielsen JB, Lester A, Friis-Møller A, Schønning K (2011) A multiplex, internally controlled real-time PCR assay for detection of toxigenic Clostridium difficile and identification of hypervirulent strain 027/ST-1. Eur J Clin Microbiol Infect Dis 31(6):1073–1079
de Boer RF, Wijma JJ, Schuurman T, Moedt J, Dijk-Alberts BG, Ott A et al (2010) Evaluation of a rapid molecular screening approach for the detection of toxigenic Clostridium difficile in general and subsequent identification of the tcdC Delta117 mutation in human stools. J Microbiol Methods 83(1):59–65
Niesters HG (2001) Quantitation of viral load using real-time amplification techniques. Methods 25(4):419–429
Pepe MS (2003) Comparing binary tests and regression analysis. In: The statistical evaluation of medical tests for classification and prediction. Oxford University Press, New York, pp 35–65
Wilcox MH, Planche T, Fang FC, Gilligan P (2010) What is the current role of algorithmic approaches for diagnosis of Clostridium difficile infection? J Clin Microbiol 48(12):4347–4353
Planche TD, Davies KA, Coen PG, Finney JM, Monahan IM, Morris KA et al (2013) Differences in outcome according to Clostridium difficile testing method: a prospective multicentre diagnostic validation study of C difficile infection. Lancet Infect Dis 13(11):936–945
Berry N, Sewell B, Jafri S, Puli C, Vagia S, Lewis AM et al (2014) Real-time polymerase chain reaction correlates well with clinical diagnosis of Clostridium difficile infection. J Hosp Infect 87(2):109–114
Hink T, Burnham CA, Dubberke ER (2013) A systematic evaluation of methods to optimize culture-based recovery of Clostridium difficile from stool specimens. Anaerobe 19:39–43
Hill KA, Collins J, Wilson L, Perry JD, Gould FK (2013) Comparison of two selective media for the recovery of Clostridium difficile from environmental surfaces. J Hosp Infect 83(2):164–166
Perry JD, Asir K, Halimi D, Orenga S, Dale J, Payne M et al (2010) Evaluation of a chromogenic culture medium for isolation of Clostridium difficile within 24 hours. J Clin Microbiol 48(11):3852–3858
Peterson LR, Mehta MS, Patel PA, Hacek DM, Harazin M, Nagwekar PP et al (2011) Laboratory testing for Clostridium difficile infection: light at the end of the tunnel. Am J Clin Pathol 136(3):372–380
van den Berg RJ, Ameen HA, Furusawa T, Claas EC, van der Vorm ER, Kuijper EJ (2005) Coexistence of multiple PCR-ribotype strains of Clostridium difficile in faecal samples limits epidemiological studies. J Med Microbiol 54(Pt 2):173–179
Hell M, Permoser M, Chmelizek G, Kern JM, Maass M, Huhulescu S et al (2011) Clostridium difficile infection: monoclonal or polyclonal genesis? Infection 39(5):461–465
Tenover FC, Novak-Weekley S, Woods CW, Peterson LR, Davis T, Schreckenberger P et al (2010) Impact of strain type on detection of toxigenic Clostridium difficile: comparison of molecular diagnostic and enzyme immunoassay approaches. J Clin Microbiol 48(10):3719–3724
René P, Frenette CP, Schiller I, Dendukuri N, Brassard P, Fenn S et al (2012) Comparison of eight commercial enzyme immunoassays for the detection of Clostridium difficile from stool samples and effect of strain type. Diagn Microbiol Infect Dis 73(1):94–96
Goldenberg SD, Gumban M, Hall A, Patel A, French GL (2011) Lack of effect of strain type on detection of toxigenic Clostridium difficile by glutamate dehydrogenase and polymerase chain reaction. Diagn Microbiol Infect Dis 70(3):417–419
Babady NE, Stiles J, Ruggiero P, Khosa P, Huang D, Shuptar S et al (2010) Evaluation of the Cepheid Xpert Clostridium difficile Epi assay for diagnosis of Clostridium difficile infection and typing of the NAP1 strain at a cancer hospital. J Clin Microbiol 48(12):4519–4524
Pancholi P, Kelly C, Raczkowski M, Balada-Llasat JM (2012) Detection of toxigenic Clostridium difficile: comparison of the cell culture neutralization, Xpert C. difficile, Xpert C. difficile/Epi, and Illumigene C. difficile assays. J Clin Microbiol 50(4):1331–1335
Kuijper EJ, Coignard B, Tüll P; ESCMID Study Group for Clostridium difficile; EU Member States; European Centre for Disease Prevention and Control (2006) Emergence of Clostridium difficile-associated disease in North America and Europe. Clin Microbiol Infect 12(Suppl 6):2–18
Bauer MP, Notermans DW, van Benthem BH, Brazier JS, Wilcox MH, Rupnik M et al (2011) Clostridium difficile infection in Europe: a hospital-based survey. Lancet 377(9759):63–73
Persson S, de Boer RF, Kooistra-Smid AM, Olsen KE (2011) Five commercial DNA extraction systems tested and compared on a stool sample collection. Diagn Microbiol Infect Dis 69(3):240–244
Monteiro L, Bonnemaison D, Vekris A, Petry KG, Bonnet J, Vidal R et al (1997) Complex polysaccharides as PCR inhibitors in feces: Helicobacter pylori model. J Clin Microbiol 35(4):995–998
Akane A, Matsubara K, Nakamura H, Takahashi S, Kimura K (1994) Identification of the heme compound copurified with deoxyribonucleic acid (DNA) from bloodstains, a major inhibitor of polymerase chain reaction (PCR) amplification. J Forensic Sci 39(2):362–372
Abu Al-Soud W, Rådström P (2000) Effects of amplification facilitators on diagnostic PCR in the presence of blood, feces, and meat. J Clin Microbiol 38(12):4463–4470
Kaneko H, Kawana T, Fukushima E, Suzutani T (2007) Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances. J Biochem Biophys Methods 70(3):499–501
Nixon G, Garson JA, Grant P, Nastouli E, Foy CA, Huggett JF (2014) Comparative study of sensitivity, linearity, and resistance to inhibition of digital and nondigital polymerase chain reaction and loop mediated isothermal amplification assays for quantification of human cytomegalovirus. Anal Chem 86(9):4387–4394
Acknowledgements
We gratefully acknowledge the assistance of the microbiology staff in the faeces laboratory at the Department of Clinical Microbiology, Slagelse Hospital and at Statens Serum Institut, Copenhagen, Denmark, and all the patients who took part in the study. We thank Diagen and Simoco Diagnostics for providing the platforms and kits. The present study was supported by grants from the Region of Zealand’s Health Research Foundation and the Hospital South’s Local Research Foundation.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 105 kb)
Rights and permissions
About this article

Cite this article
Jensen, M.B.F., Olsen, K.E.P., Nielsen, X.C. et al. Diagnosis of Clostridium difficile: real-time PCR detection of toxin genes in faecal samples is more sensitive compared to toxigenic culture. Eur J Clin Microbiol Infect Dis 34, 727–736 (2015). https://doi.org/10.1007/s10096-014-2284-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-014-2284-7