
Abstract
Aim
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) is an inherited rare disease affecting young adults. We present the clinical, imaging, and neuropathological results of our case series, emphasizing biopsy histology combined with clinical information will increase the accuracy of early diagnosis.
Methods
In total, 4 females and 2 male ALSP patients with onset at ages 24–45 years were enrolled. Clinical manifestations, neuroimaging, and histopathology as well as gene mutation were analyzed and compared with literature.
Results
Clinical manifestations include cognitive decline with/without psycho-behavior problems and movement disorders including paralysis, hemiplegia, parkinsonism, and pyramidal tract injury, as well as dysarthria, dysphagia, and sensory disturbances. MRI showed multiple periventricular and subcortical white matter lesions, involving the corpus callosum, with no enhancement, but with persistent hyperintensity on diffuse-weighted imaging. Histology showed widespread white matter damage and pale stain, especially destroyed axons with spheroids and funicular axons which were stained with neurofilament and ubiquitin. Foamy and pigmented macrophages were another typical change. CSF1R mutation was found in 4 of them. All of the patients were misdiagnosed and treated for a long time for multiple sclerosis, cerebral infarction, normal pressure hydrocephalus, etc.
Conclusion
ALSP will cause rapidly progressing dementia with/without movement disorders in young adults. The definite diagnosis should be based on a comprehensive analysis of clinical manifestations, and neuroimaging, histology, and genetic results. Early biopsy will add to the accuracy of the diagnosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) was regarded as a distinct disease entity after the identification of pathogenic gene CSF1R in 2013 [1]. It includes a disease spectrum defined by clinical and pathological features previously, namely hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) and pigmented orthochromatic leukodystrophy (POLD). Both are members of orthochromatic leukodystrophy: HDLS was characterized by widespread degeneration and loss of myelin sheaths and axons, and abundant neuroaxonal spheroids, while POLD was characterized by pigmented macrophages in a background of widespread myelin loss and axon damage [2].
The clinical spectrum of ALSP includes cognitive disorder, behavior or psychiatric symptoms, and movement disorders, as well as seizure, ataxia, parkinsonism, and gait disturbance [3, 4]. Dementia and frontal release signs are predominantly found because of frontal-predominant white matter involvement, differentiated from bvFTD [5]. The characteristic MRI changes are patchy or confluent, symmetric or asymmetric white matter lesions with frontal periventricular predominance atrophy of the genu of the corpus callosum and enlargement of the lateral ventricle. Persistent diffuse-weighted imaging (DWI) signal increase without gadolinium enhancement is also important [4, 6]. Although CSF1R mutation is pathogenic, more and more mutation-negative cases were reported and AARS2 mutation caused a similar phenotype, so genetic diagnosis was still not specific [7]. Characteristic histology manifestations, therefore, will still be emphasized [8].
Here, we present the clinical, imaging, and neuropathological results of our series from leukoencephalopathy and dementia clinics. We emphasize ALSP should be taken into account in differential diagnosis of young dementia and biopsy histology will add to the accuracy of early diagnosis.
Materials and methods
Clinical data
Six cases with biopsy-proven ALSP were included in the study. Clinical history including age onset, disease duration, family history, symptoms, and physical examinations was recorded. Laboratory results were collected, including serum analysis for biochemistry (liver function, kidney function, cholesterol, homocysteine, et al.), autoimmune antibodies (anti-nuclear antibodies, extractable nuclear antigen, anti-neutrophil cytoplasmic antibodies, aquaporin-4 antibody), endocrine and metabolism indexes (thyroid function, alpha-galactosidase, beta-galactosidase, galactocerebrosides, arylsulfatase, hexosaminidase), and CSF indexes (cell count, protein, glucose, chloride, oligoclonal band, bacterial culture, and antibodies/PCR of viruses, cytology). Otherwise, the clinical course and treatment history of the patients were also collected.
MRI studies
MRI examinations were performed using a 3-T MRI scanner (Avanto, Siemens, Erlangen, Germany). Axial T1-weighted, T2-weighted, T2-flair, and DWI were performed and corresponding decreased apparent diffusion coefficient (ADC) was measured. Sagittal and coronal imaging, and contrast-enhanced studies were performed.
Histopathological examination
Four-micrometer-thick paraffin sections were stained with routine stains (hematoxylin and eosin). Immunohistochemistry illustrated T and B cells CD3/CD20 (Leica), microglia/macrophages CD68 (DAKO), neurofilament protein NF (DAKO), neurons Neun (Abcam), astrocytes GFAP (Abcam), and ubiquitin (Abcam). PAS was stained in some of the cases. Photomicrographs were taken with the LEICA DM2500 microscope and a digital camera (LEICA, Germany).
Molecular genetic analysis
Written informed consent for genetic analysis was obtained from all investigated members or their legal representatives. Genomic DNA was extracted from fresh peripheral blood leukocytes. Whole exon sequencing using “next-generation” sequencing technology was performed on IlluminaHiseq (Illumina, USA), which was verified by Sanger sequencing.
Results
Clinical information
Among the 6 cases, female:male was 4:2. The age onset was from 24 to 45 years, an average of 38 ± 7.5 years, all in young adult period. The duration from onset to diagnosis was from 9 to 48 months, for an average of 20.2 ± 14.0 months. Only one patient reported family history without genetic confirmation, while the rest denied any similar patients in family. The onset clinical symptoms could be divided into two main groups: generalized cognitive decline with/without psychiatric problems and focal cortical neurological deficits. As the disease progressed, symptoms could overlap and cover all manifestations of the central nervous system. The detailed symptoms of the patients are listed in Table 1. Ataxia and parkinsonism were relatively rare while epilepsy was not reported. The laboratory examinations were all unremarkable, including serum biochemistry, antibodies, inflammatory indexes, tumor biomarkers, et al. Lumbar puncture was done in all patients and CSF indexes were all negative, including oligoclonal band and cytology. Clinical diagnosis was inflammatory demyelinating disease in cases 1, 2, 3, and 5, cerebral infarction in case 4, and hydrocephalus in case 6. Steroids, anti-infarction, and CSF drainage were given respectively without efficacy.
Neuroimaging manifestations
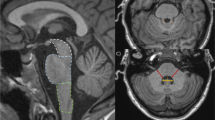
The neuroimaging manifestations are summarized in Table 2. MRI showed white matter lesions prominently in all patients, with widespread periventricular and sub cortical white matter involvement, except for case 2 which showed patchy periventricular localization. Four patients had frontal predominance with frontal lobe atrophy while the 2 had parietal occipital predominance with enlargement of occipital horns of the lateral ventricle. Corpus callosum (5/6) and pyramidal tract (4/6) involvements were seen in most of them. However, brain stem lesions were not common (2/6). Persistent DWI high signal was seen in all patients and almost in all lesions, lasting more than 1 year for the longest time. All lesions were distributed asymmetrically, without enhancement and U-fiber involvement. Calcification was found in 2 of them. Illustrations of MRI are shown in Fig. 1.

MRI of patients. a–c Case 1: confluent periventricular white matter lesions with subcortical involvement and DWI hyperintensity (T2, FLAIR, DWI). d–f Case 2: frontal predominant patchy white matter lesions with DWI hyperintensity (T1, T2, DWI). g, h Case 3: frontal periventricular white matter lesions with corpus callosum involvement and calcification (FLAIR, CT). i–l Case 4: parietal occipital predominant white matter lesions with no enhancement and DWI hyperintensity, and brain stem lesions (FLAIR, T1+C, DWI,FLAIR). m, n Case 5:periventricular lesions with DWI hyperintensity and pyramidal tract (internal capsule) involvement (FLAIR, DWI). o, p Case 6: confluent white matter lesions with corpus callosum involvement and ventricle enlargement, no enhancement (FLAIR, T1+C)
Neuropathological investigations
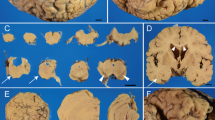
Periventricular white matter tissues with high DWI signal were biopsied in all patients. Illustrations of histology are shown in Fig. 2. Microscopically, white matter was wildly damaged and oligodendrocytes massively reduced. In severely affected areas, axon and myelin were almost dismissed, with a spongiform pattern and patchy astrogliosis. However, in mildly affected areas, axon breakdown and derangement were more obvious, while myelin was relatively preserved. Spheroid and thickened (or funicular) axons were easily found in mildly affected areas. Neither necrosis nor inflammatory lymphocyte infiltration and vascular changes were found. Scattered macrophages were found distributed in the tissue, usually single without gathering and vascular surrounding. Plasma of macrophages was enriched with axon breakdown which was stained with PAS, so-called pigmented macrophages. On immunohistochemical stain, GFAP showed extensive gliosis, and some of the reactive astrocytes had massive cytoplasm and projections. Seldom were CD3- or CD20-positive lymphocytes seen. Scattered macrophages were CD68 positive. NF showed extensive axon damage and better outlined spheroids and cord like axons. Ubiquitin was stained in 3 of the patients (cases 2, 3, and 4), showing positive spheroids.

Histology of patients. a–c Case 1: diffuse white matter damage with funicular, spheroid axons and pigmented macrophages, myelin relatively kept in mild affected area (NFX200, CD68 X200, LFB X100). d–f Case 2: axon damage and spheroid axons without vasculature inflammation, severe gliosis in widespread spongiform white matter background (HE × 200, HE × 200, NF × 100). g, h Case 3: spheroid axons and scattered pigmented macrophages (HE × 400, CD68 × 200). i–m Case 4: spheroid, funicular axons and pigmented macrophages in severely destroyed white matter; PAS-positive granules in macrophages and Ub-positive stain of spheroid axons (HE × 100, HE × 200, NF × 200, PAS × 200, Ub × 200). n, o Case 5: spheroid, funicular axons and pigmented macrophages (NF × 200, CD68 × 200). p Case 6: severe destruction and derangement of axons (NF × 100)
CSF 1R gene analysis
Case 1 refused a gene analysis and no CSF1R mutation was found in case 3. In case 2, delCTC mutation was detected in CSF1R gene. In case 4, a new deletion mutation (c.2546-2548delTCT) was found that did not exist in both parents. In case 5, c.2381T>C, p.I794T was detected. In case 6, c.2563C>A missense mutation was detected.
Discussion
As a subgroup of adult-onset leukodystrophy, ALSP was first described as a clinical entity, then a pathological entity, a genetic entity, and now a clinical-pathological-genetic unity. As typical pathologic and genetic features were found, more and more cases and families were reported all around the world [9,10,11]. Konno et al. raised diagnostic criteria based on the clinical characteristics. Age onset, typical clinical symptoms, and typical imaging features were included in core features. Neuropathological findings were included in supporting features. However, other causes of leukoencephalopathy should be excluded [12].
The clinical features of ALSP covered a wide spectrum of the central nervous system. Focal neurological deficits involving pyramidal signs and motor disorders along with periventricular white matter lesions always mimicked demyelinating disease especially multiple sclerosis [13, 14]. Besides, onset of cognitive decline would make the diagnosis of neurodegenerative disease such as early-onset Alzheimer’s disease, frontal temporal lobe degeneration, et al. [5, 15]. Parkinsonism and movement disorders should be differentiated [15, 16]. At last, white matter lesions should also be differentiated with central nervous system vasculitis, small vessel disease such as CADASIL, and other hereditary leukoencephalopathy and leukodystrophy [17]. In our case series, the onset age and disease progression course were similar to literature. Cognitive decline, psychiatric symptoms, pyramidal signs, and motor deficits were most frequently described in literature. However, ataxia and epilepsy were not found. All of our patients were misdiagnosed for a long time: 4 with multiple sclerosis and steroid was given with no clinical benefit. Persistent DWI hyperintensity with no enhancement and spot-like calcification were different from MS lesions. Many of them were treated as cerebral infarction in rural hospitals for DWI hyperintensity. It should be specially mentioned that case 6 was misdiagnosed as normal pressure hydrocephalus and tap test was done with no improvement.
Spheroid axons and pigmented macrophages were key pathological features of ALSP. Pyramidal tract and corpus callosum were usually affected. In white matter, diffuse loss of axons and myelin, numerous spheroids, and intense astrogliosis were seen. Cortical lamination was usually kept and neuron loss was not usually found. However, a few balloon-like neurons could be seen [18]. The cytoplasm of pigmented macrophages was positively stained by PAS, Sudan III, Berlin blue, and ubiquitin [18]. Besides, it was reported that enlarged unmyelinated axon was also found in skin tissue, which meant peripheral nervous system involvement [19]. As we know, axon damage and spheroid axons were not specific, and may be caused by lots of pathophysiologic process such as Wallerian degeneration, traumatic axon damage, and other secondary reactions. However, the widespread distribution and frequency were specific. Also, the scattered distribution of macrophages was distinct without perivascular accumulation, which was not usually seen in other metabolic leukoencephalopathy and demyelinating disorders. Autopsy material from asymptomatic member of a Japanese family showed patchy axonal degeneration and myelin loss, predominantly in the subcortical white matter; however, pigmented microglia was distributed diffusely throughout the cerebral white matter, suggesting microglia pathophysiologic changes came earlier [20]. Axon loss was a pathologic result and progressed from patchy localization to widespread diffusion. Based on these, the Japanese researchers raised a pathological staging system of ALSP, mainly considering distribution of axon damage and atrophy [21]. In our series, all pathological features were in the advanced stage, for delayed clinical diagnosis. We found typical spheroid axons, and thickened axons in longitudinal section which we called funicular axons. The distribution and morphologic features of pigmented macrophages were all as described in literature.
CSF1R is a tyrosine kinase receptor expressed on the surface of microglia, which was found to be the pathogenic mutation of ALSP [1]. Konno et al. studied 90 families with ALSP and found 58 mutations located in the tyrosine kinase domain of CSF1R [22]. However, 40% of the patients have no family history. The detailed pathogenesis of CSF1R mutation was unknown. TREM2, another gene involved in activation of microglias, was found to cause Nasu–Hakola disease while mutated. Nasu–Hakola disease is characterized by marked degeneration of cerebral white matter with spheroids, bone fracture, and peculiar membranous structures in the bone marrow and adipose tissue [21]. Besides, mutations in the alanyl-tRNA synthetase 2 (AARS2) gene causing ovario-leukodystrophy were also reported to cause similar clinical and pathological changes as ALSP [7, 23, 24]. In our series, most of them were sporadic without family history and CSF1R mutations were found in 4 of them. In case 4, no CSF1R mutation was found, suggesting other underlying genetic mechanisms. Indeed, as a clinical pathological nomenclature, ALSP is a genetic heterogeneous entity.
In conclusion, ALSP represents a subgroup of adult-onset leukoencephalopathy with distinct histological characteristics, but with a wide spectrum of clinical manifestations and genetic heterogeneity. We should take ALSP into account with diagnosis of rapid progressing dementia with/without movement disorders in young adults. Asymmetrical white matter lesions with persistent DWI hyperintensity without contrast as well as corpus callosum involvement are characteristic MRI features. Spheroid or funicular axons and pigmented macrophages are typical histology changes. However, secondary axon changes should be ruled out on biopsy. The definite diagnosis should be based on a comprehensive analysis of clinical manifestations, and neuroimaging, histology, and genetic results. To avoid misdiagnosis and delayed diagnosis, we suggest early biopsy in these patients to get rid of side effects and costs of wrong treatment and help them keep a relatively high quality of life.
References
Nicholson AM, Baker AM, Finch NA, Rutherford NJ, Wider C, Graff-Radford NR et al (2013) CSF1R mutations link POLD and HDLS as a single disease entity. Neurology 80:1033–1040
Marotti JD, Tobias S, Fratkin JD, Powers JM, Rhodes CH (2004) Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol 107:481–488
Terada S, Ishizu H, Yokota O, Ishihara T, Nakashima H, Kugo A et al (2004) An autopsy case of hereditary diffuse leukoencephalopathy with spheroids, clinically suspected of Alzheimer’s disease. Acta Neuropathol 108:538–545
Wider C, Van Gerpen JA, DeArmond S, Shuster EA, Dickson DW, Wszolek ZK (2009) Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity? Neurology 72:1953–1959
Wong JC, Chow TW, Hazrati LN (2011) Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia can present as frontotemporal dementia syndrome. Dement Geriatr Cogn Disord 32:150–158
Maillart E, Rousseau A, Galanaud D, Gray F, Polivka M, Labauge P, Hauw JJ, Lyon-Caen O, Gout O, Sedel F (2009) Rapid onset frontal leukodystrophy with decreased diffusion coefficient and neuroaxonal spheroids. J Neurol 256:1649–1654
Lynch DS, Zhang WJ, Lakshmanan R, Kinsella JA, Uzun GA, Karbay M, Tüfekçioglu Z, Hanagasi H, Burke G, Foulds N, Hammans SR, Bhattacharjee A, Wilson H, Adams M, Walker M, Nicoll JA, Chataway J, Fox N, Davagnanam I, Phadke R, Houlden H (2016) Analysis of mutations in AARS2 in a series of CSF1R-negative patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol 73:1433–1439
Freeman SH, Hyman BT, Sims KB, Hedley-Whyte ET, Vossough A, Frosch MP et al (2009) Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol 19:39–47
Mendes A, Pinto M, Vieira S et al (2010) Adult-onset leukodystrophy with axonal spheroids. J Neurol Sci 297:40–45
Kleinfeld K, Mobley B, Hedera P, Wegner A, Sriram S, Pawate S (2013) Adult-onset leukoencephalopathy with neuroaxonal spheroids and pigmented glia: report of five cases and a new mutation. J Neurol 260:558–571
Kim E-J, Shin J-H, Lee JH et al (2015) Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia linked CSF1R mutation: report of four Korean cases. J Neurol Sci 349:232–238
Konno T, Yoshida K, Mizuta I, Mizuno T, Kawarai T, Tada M, Nozaki H, Ikeda SI, Onodera O, Wszolek ZK, Ikeuchi T (2018) Diagnostic criteria for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia due to CSF1R mutation. Eur J Neurol 25:142–147
Martinez-Saez E, Shah S, Costa C, Fleminger S, Connor S, Bodi I (2012) Adult onset leukodystrophy with neuroaxonal spheroids and demyelinating plaque-like lesions. Neuropathology 32:285–292
Keegan BM, Giannini C, Parisi JE, Lucchinetti CF, Boeve BF, Josephs KA (2008) Sporadic adult-onset leukoencephalopathy with neuroaxonal spheroids mimicking cerebral MS. Neurology 70:1128–1133
Ikeuchi T, Mezaki N, Miura T et al (2018) Cognitive dysfunction and symptoms of movement disorders in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Parkinsonism Relat Disord 46:S39–S41
Sundal C, Fujioka S, Van Gerpen JA et al (2013) Parkinsonian features in hereditary diffuse leukoencephalopathy with spheroids (HDLS) and CSF1R mutations. Parkinsonism Relat Disord 19:869–877
Lynch DS, Jaunmuktane Z, Sheerin U-M et al (2016) Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry 87:512–519
Itoh K, Shiga K, Shimizu K, Muranishi M, Nakagawa M, Fushiki S (2006) Autosomal dominant leukodystrophy with axonal spheroids and pigmented glia: clinical and neuropathological characteristics. Acta Neuropathol 111:39–45
Hoffmann S, Murrell J, Harms L, Miller K, Meisel A, Brosch T, Scheel M, Ghetti B, Goebel HH, Stenzel W (2014) Enlarging the nosological spectrum of hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). Brain Pathol 24:452–458
Riku Y, Ando T, Goto Y, Mano K, Iwasaki Y, Sobue G, Yoshida M (2014) Early pathologic changes in hereditary diffuse leukoencephalopathy with spheroids. J Neuropathol Exp Neurol 73:1183–1190
Oyanagi K, Kinoshita M, Suzuki-Kouyamal E et al (2017) Adult onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and Nasu–Hakola disease: lesion staging and dynamic changes of axons and microglial subsets. Brain Pathol 27:748–769
Konno T, Yoshida K, Mizuno T, Kawarai T, Tada M, Nozaki H, Ikeda SI, Nishizawa M, Onodera O, Wszolek ZK, Ikeuchi T (2017) Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur J Neurol 24:37–45
Lakshmanan R, Adams ME, Lynch DS et al (2017) Redefining the phenotype of ALSP and AARS2 mutation–related leukodystrophy. Neurol Genet 3:e135
Wang D, Yu M, Zhang W et al (2018) AARS2 compound heterozygous variants in a case of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neuropathol Exp Neurol 77:997–1000
Funding
This work was supported by Chinese Academy of Medical Sciences Innovation fund for medical sciences (2016-I2M-1-004), National Natural Science Foundation of China (81550021), 13th Five year National Key Research and Development Program of China (2016YFC1306300), and the strategic priority research program (Pilot study) “Biological Basis of Aging and Therapeutic Strategies” of the Chinese Academy of Sciences (grant XDPB10).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (PUMC ethics committee 2017006) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mao, C., Zhou, L., Zhou, L. et al. Biopsy histopathology in the diagnosis of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). Neurol Sci 41, 403–409 (2020). https://doi.org/10.1007/s10072-019-04116-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-019-04116-7