
Abstract
Very few cases of patients with myasthenia gravis (MG) who later developed amyotrophic lateral sclerosis (ALS) have been described, although some studies showed that significantly more cases than expected have ALS associated with a prior diagnosis of autoimmune diseases. Our aim was to investigate whether the association of ALS and MG was higher than expected in a population-based study and to describe the clinical features characterizing these patients. In Emilia Romagna Region of Italy, a prospective registry has been collecting all incident ALS cases since 1.1.2009. For each patient, detailed clinical information is collected by caring physicians, including comorbidities. From 1.1.2009 to 31.12.2014, 671 patients were diagnosed with ALS; five patients (0.75%) were also affected by MG. Considering Western Countries incidence rates the occurrence of both the diseases should be a really exceptional event (7.5/109), compared to our findings (1.87/107) (p < 0.01). Patients with ALS and MG had more frequently a bulbar onset and a fast progressive course. These cases of ALS after MG raise the possibility of potential shared immunological dysfunctions, which may be expression of common pathogenic mechanisms, as well as of shared disease-course modulating events.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Only a few cases of patients with myasthenia gravis (MG) who later developed amyotrophic lateral sclerosis (ALS) have been described [1], although a recent study showed that significantly more cases than expected have ALS associated with a prior diagnosis of autoimmune diseases [2].
Our aim was to investigate whether the association of ALS and MG was higher than expected in a population-based study and to describe the clinical features characterizing these patients.
Materials and methods
In Emilia Romagna region of Italy (4.5 million population), a prospective registry (ERRALS) has been collecting all the incident ALS cases since 1.1.2009 [3].
Caring physicians collect a detailed phenotypic profile of each ALS patient, including demographic and clinical data among which clinical phenotype, presence of dementia, extrapiramidal signs, or any other pathological condition, the use of enteral nutrition and non-invasive or invasive ventilatory support, and date and cause of death. These data have been included into an electronic database available through a dedicated internet website to ERRALS participants; regular supervision on the data has been performed by the coordinating centre (Modena).
Analysis for comorbidities has been performed considering this data source. Clinical features of cases with ALS and MG were collected from caring physicians.
Descriptive statistics were performed using Student T test and Chi-square test as appropriate. The probability of the occurrence of the two diseases was calculated considering incidence rates.
Results
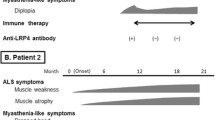
Among 671 patients diagnosed with ALS through 6 years, five patients (0.75%) were also affected by MG. Their clinical and demographic characteristics are described in Table 1.
In all the cases, the diagnosis of MG preceded the diagnosis of ALS, with a variable delay. MG symptoms at onset were set in the ocular district for all the cases, and were associated with bulbar localization in two cases; in three cases, there was generalized fatigability. Bulbar-onset ALS represented the 60% of cases with ALS and MG, with a predominance of bulbar phenotype (p < 0.01).
Patients diagnosed with ALS after MG were slightly older and with a short ALS diagnostic delay compared to other patients with ALS (Table 2). Patients diagnosed with both the diseases underwent NIV more frequently than the general ALS cohort (p < 0.01) and had a rapidly progressing disease course.
Considering Western Countries incidence rates (ALS: 2–3/100,000; MG: 30/100,000) [3, 4], a chance occurrence of the two disorders should be a really exceptional event.
In fact, in Emilia Romagna Region, annual ALS incidence rates in the period 2009–2014 varied from 2.41 to 2.69/100,000 with a mean ALS incidence in the region of 2.52/100,000. Considering the number of expected MG cases based on Western Countries rates, we should expect 7.55/109 cases of ALS with MG, whereas we found 1.87/107 (p < 0.01).
Discussion
With limitations related to the relatively small size of our studied group, this study highlights some peculiar features of ALS cases with MG. The frequent bulbar onset of ALS symptoms may explain the lower survival due to nutritional deficiency and/or an earlier progression to brainstem respiratory centres due to proximity of affected areas, as confirmed by the early onset of respiratory symptoms in these five patients.
In a retrospective series of six cases with a concomitant diagnosis of ALS and MG collected among a French cohort of 4757 patients followed at 18 reference centers for ALS in a 12-year period, Amador Del Mar et al. [1] described three cases with initial MG and subsequent ALS development and three cases of ALS with subsequent diagnosis of MG. We described only patients with MG who later developed ALS. This may be due to the difficulty to think about MG during the course of a devastating disease, such as ALS, as stated by the authors [1]. Accordingly, with their data, patients who developed ALS after MG were older and had a frequent bulbar onset of ALS symptoms (60%) compared to patients with ALS alone (reported bulbar-onset frequency in ALS: 30–40% of cases) [3]. These data may explain also the shortened diagnostic latency, which is a surrogate marker of disease progression. Survival is not reported in the study of Amador Del Mar et al. [1], and it would be interesting to make a comparison with their data.
In our cohort, diagnoses were confirmed after revision of cases with the caring neurologists of the ALS Centres in Emilia Romagna Region, and were based mainly on clinical and neurophysiological (repetitive nerve stimulation and/or single fibre EMG) data and on treatment response; three of our cases were seronegative MG, but with an enduring effect of acetylcholinesterase (AchE) inhibitors on ocular symptoms and exhaustibility, antecedent to ALS diagnosis. This is another difference with the French cohort [1], which could be explained by the small number of available cases, and suggests that further studies should be performed.
From an epidemiological point of view, considering Western Countries incidence rates (ALS: 2–3/100,000; MG: 30/100,000) [3, 4], a chance occurrence of the two disorders should be a really exceptional event (7.5/109), compared to our data (1.87/107) (p < 0.01).
The prospective, population-based design of our study can explain this difference with recent reports [1, 2], in which the retrospective nature may lead to some loss of cases. This unusual association can be “a by chance association” as well, but these cases raise the question whether the development of ALS after MG can represent an association based on shared pathogenic mechanisms.
Apart from antibodies described in MG and ALS, among which LRP4 antibodies that are crucial in the development and function of motor neurons and neuromuscular junction [5, 6], a possible link between the two diseases may be represented by immunoregulatory defects of regulatory T lymphocytes (Treg) [7, 8]. Treg are CD4 cells coexpressing CD25 and FoxP3 (a transcription factor essential for their regulatory function) and playing an important role in autoimmune and immune responses. Tregs downregulate pro-inflammatory cytokines production, secrete anti-inflammatory cytokines (IL4, IL10, and IL13), and neurotrophic factors, transform a Th1 to Th2 response, and attenuate toxic microglial responses. Tregs have been shown to directly differentiate macrophages from M1 to M2 states [9]. While in MG patients, the number of Tregs is unchanged, but they have a severe defect in their suppressive function [7], evidences for a “benign” role of Treg in ALS come from in vitro and in vivo studies showing a role of Tregs in promoting MN survival by suppressing M1 activation, and in reducing the release of reactive oxygen species [10]. In mSOD1 mice, increased Tregs and M2 microglia were associated with the stable phase of disease, suggesting a shift from protection to toxicity [11]. In ALS patients, Tregs percentage in the blood inversely correlated with ALS progression rate; in addition, Treg numbers and FoxP3 expression decreased with faster ALS progression [11]. These data were confirmed in post-mortem studies [8].
Nevertheless, because immunological investigations in MG and ALS patients are performed when the disease is already well established, it remains unclear whether Tregs dysfunction is a causal event or a result of perturbations of the immune system occurs during disease development because of the inflammatory environment [7].
In conclusion, we think that the co-occurrence of ALS and MG deserves further studies for a better diagnostic and phenotypical characterization and raise the possibility of potential shared immunological dysfunctions, which may be expression of common pathogenic mechanisms, as well as of shared disease-course modulating events.
References
Del Mar Amador M, Vandenberghe N, Berhoune N et al (2016) Unusual association of amyotrophic lateral sclerosis and myasthenia gravis: a dysregulation of the adaptive immune system? Neuromuscul Disord 26:342–346. doi:10.1016/j.nmd.2016.03.004
Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ (2013) Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology 81:1222–1225. doi:10.1212/WNL.0b013e3182a6cc13
Mandrioli J, Biguzzi S, Guidi C et al (2014) Epidemiology of amyotrophic lateral sclerosis in Emilia Romagna Region (Italy): a population based study. Amyotroph Lateral Scler Frontotempor Degener 15:262–268. doi:10.3109/21678421.2013.865752
McGrogan A, Sneddon S, de Vries CS (2010) The incidence of myasthenia gravis: a systematic literature review. Neuroepidemiology 43:171–183. doi:10.1159/000279334
Gotaas HT, Skeie GO, Gilhus NE (2016) Myasthenia gravis and amyotrophic lateral sclerosis: a pathogenic overlap. Neuromuscul Disord 26:337–341. doi:10.1016/j.nmd.2016.03.003
Tzartos JS, Zisimopoulou P, Rentzos M et al (2014) LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol 1:80–87. doi:10.1002/acn3.26
Berrih-Aknin S, Le Panse R (2014) Myasthenia gravis: a comprehensive review of immune dysregulation and etiological mechanisms. J Autoimmun 52:90–100. doi:10.1016/j.jaut.2013.12.011
Henkel JS, Beers DR, Wen S et al (2013) Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med 5:64–79. doi:10.1002/emmm.201201544
Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS (2007) CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. PNAS USA 104:19446–19451
Zhao W, Xie W, Xiao Q, Beers DR, Appel SH (2006) Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia. J Neurochem 99:1176–1187
Beers DR, Henkel JS, Zhao W et al (2011) Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 134:1293–1314. doi:10.1093/brain/awr074
Acknowledgements
The ALS Registry is supported by a Grant from the Emilia Romagna Regional Health Authority.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Rights and permissions
About this article

Cite this article
de Pasqua, S., Cavallieri, F., D’Angelo, R. et al. Amyotrophic lateral sclerosis and myasthenia gravis: association or chance occurrence?. Neurol Sci 38, 441–444 (2017). https://doi.org/10.1007/s10072-016-2787-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-016-2787-3