
Abstract
In this study, we aimed to explore the expression levels of JAK2 and PTPRC in peripheral blood mononuclear cells (PBMCs) from SLE patients and controls, detect the effects of SLE activity on genes mRNA expression, and find the association between genes mRNA expression and clinical manifestations of patients. We performed quantitative real-time PCR (qRT-PCR) to test differences in the expression levels of JAK2 and PTPRC in PBMCs extracted from 135 patients with SLE and 130 healthy controls. Furthermore, we detected the regulatory effect of SNPs on gene expression by expression quantitative trait loci (eQTL). We also tested whether the genes mRNA expression was affected with the SLE activity and analyzed the relationship between genes mRNA expression and clinical manifestations of patients. The mRNA expression levels of JAK2 in SLE patients were significantly higher than those in healthy controls (P = 0.005), and PTPRC mRNA expression levels were significantly decreased (P < 0.001). However, no other statistical significance was detected. We found that the elevated JAK2 mRNA expression and the decreased PTPRC mRNA expression may play suggestive roles in the pathogenesis of SLE.
Key Points • The JAK2 mRNA expression levels in SLE patients were significantly higher than those in healthy controls. • The PTPRC mRNA expression levels in SLE were decreased. • JAK2 and PTPRC mRNA expression may play suggestive roles in the pathogenesis of SLE. |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disorder that presents a variety of complex clinical manifestations and affects multiple organ systems. The prevalence of SLE ranges from approximately 30 to 50 per 100,000 individuals [1]. Although the precise etiology of SLE is not explicit, epidemiological investigation indicated that SLE has a definite hereditary trend and follows the polygenic model of inheritance [2]. During the past decade, genome-wide association studies (GWASs) in multiple ethnic populations have revealed more than 100 common single-nucleotide polymorphisms (SNPs) that are strongly associated (P < 5 × 10−8) with SLE [3]. We revealed in a cross-racial GWAS that ten novel loci—ZFP90, IKBKE, LBH, LPP, ATXNI, BACH2, GTF2I, RNASEHZC, JAK2, and PTPRC—are highly correlated with SLE [4]. The PTPRC gene encodes a member of the protein tyrosine phosphatase (PTP) family known as CD45, which is a critical regulator of antigen receptor signaling in T and B cells, suppresses JAK kinases and negatively regulates cytokine receptor signaling [5]. The JAK2 gene is a member of the Janus kinase (JAK) family, which includes JAK1, JAK2, JAK3, and Tyk2; these proteins form nonreceptor protein tyrosine kinases (PTKs) that are vital for cell survival, growth, development, and immune cell differentiation. Additionally, PTKs are the most integral part of the JAK/STAT signaling pathway [6]. It has been reported that dysregulation of the JAK/STAT signaling pathway may be involved in SLE progression [7]. Based on these data, we examined the mRNA expression levels of JAK2 and PTPRC in peripheral blood mononuclear cells (PBMCs) from patients with SLE and from healthy control subjects and explored whether JAK2 mRNA expression was associated with the single-nucleotide polymorphism (SNP) rs1887428. Meanwhile, we tested whether the gene mRNA expression was affected with the SLE disease activity index (SLEDAI) and analyzed the relationship between genes mRNA expression and clinical manifestations of SLE patients.
Methods
Subjects
A total of 135 patients and 130 healthy control subjects were enrolled in this study. All subjects were recruited from the First Affiliated Hospital of Anhui Medical University, Hefei, Anhui Province, China. All participants were of self-identified Han Chinese ancestry. The cases were diagnosed by at least two experienced physicians using the revised American College of Rheumatology SLE classification criteria [8], and the clinical phenotypes of the patients were documented at a full clinical check-up. Additional demographic information was obtained for each subject through a structured questionnaire. The clinical manifestations were assessed based on the American College of Rheumatology 1997 criteria for SLE. However, some clinical manifestations are not included in the 11 criteria, such as cardiac involvement and lung involvement. In this study, all the other causes result in lung or cardiac diseases should be excluded, then lung or cardiac involvement can be defined. The lung involvement includes pulmonary hypertension, pulmonary hemorrhage, pulmonary interstitial fibrosis, pneumonia, pulmonary infarction, and lung atrophy. The cardiac involvement includes myocarditis, arrhythmia, endocarditis, and coronary vascular. The SLEDAI score [9] was evaluated for each patient according to the clinical manifestations which were collected at the time of blood extracted. And a SLEDAI score ≥ 10 was defined as active SLE; otherwise, the inactive SLE was defined. The control subjects were clinically confirmed to be free of SLE and did not have any family history of SLE or any other autoimmune disease. Written informed consent was obtained from each participant. This study was approved by the Institutional Review Board of Anhui Medical University, China and was conducted according to the principles set by the Declaration of Helsinki.
Total RNA extraction
Whole blood (5 ml) was collected from all subjects in EDTA collection tubes and diluted with a 2× volume of phosphate-buffered saline (PBS). A fresh 30 ml tube was filled with 5 ml of Ficoll-Paque™ Plus (GE Healthcare), and 15 ml of the blood dilution was carefully added on top of the Ficoll-Paque. Then, the mixture was centrifuged for 25 min at 400×g at 18 °C. The cloudy-looking phase containing PBMCs was collected and centrifuged for 10 min at 400×g at 4 °C. Finally, the PBMC suspension was collected after the supernatant was decanted. TRIzol Reagent was added to extract total RNA from PBMCs. The purity and quality of RNA was measured with a NanoDrop 2000 Spectrophotometer (Thermo Scientific). The ratio of total RNA samples (ranging from 1.8 to 2.0) was used to calculate the concentration and purity prior to reverse transcription.
qRT-PCR analysis
After quantification, 400 ng of total RNA was subjected to reverse transcription using a PrimeScript RT reagent Kit (Takara). All experiments were performed in duplicate on a ViiA 7 Real-Time PCR system (Applied Biosystems) for 40 cycles of denaturation at 95 °C for 5 s, annealing and extension at 60 °C for 30 s. The endogenous expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as an internal control primer). The reactions were completed in a 5-μl reaction system containing 0.2 μl of primers (JAK2 primers: forward 5′-TCTGGGGAGTATGTTGCAGAA-3′, reverse 5′-AGACATGGTTGGGTGGATACC-3′; PTPRC primers: forward 5′-ATGATGGCAGGGTCCTTCATGT-3′, reverse 5′-AGCTTCCTCAGCTAATGATTGGACA-3′; GAPDH primers: forward 5′-CTCCTCCTGTTCGACAGTCA-3′, reverse 5′-CAATACGACCAAATCCGTTG-3′). The relative expression levels were calculated via the 2−ΔΔCt method [10].
Genotyping
Blood DNA was extracted using FlexiGene DNA Kits (QIAGEN, Germany). The SNP rs1887428 was genotyped using an improved multiplex ligation detection reaction (iMLDR) technique developed by Genesky Biotechnologies Inc. (Shanghai, China). Approximately 20 ng/μl of genomic DNA was used to genotype each sample. According to the manufacturer’s instructions, the genomic DNA was diluted to a working concentration of 5–10 ng/μl for genotyping studies. First, the sample DNA was amplified by multiplex PCR, and the resulting PCR products were then used for locus-specific single-base extension reaction. The samples were analyzed on an ABI 3730XL DNA sequencer (Applied Biosystems), and the data were analyzed using GeneMapper software (version 4.1, Applied Biosystems). Finally, 135 patients were successfully genotyped.
Statistical analysis
Data were analyzed using GraphPad Prism (version 7.04) and SPSS version 24.0. The different mRNA expression levels of JAK2 and PTPRC in SLE patients and healthy subjects were compared by the nonparametric Mann–Whitney rank sum test. The correlation of genotype and JAK2 mRNA expression levels in PBMCs was calculated by one-way analysis of variance. Two-sample rank-sum tests were performed to assess the effect of SLEDAI on two genes mRNA expression and analyze the association between two genes mRNA expression and clinical manifestations of SLE patients. P values (two-tailed) less than 0.05 were considered statistically significant, but the Bonferroni-corrected test was used in analyzing the relationship between two genes mRNA expression and clinical manifestations (significant criteria P < 0.05/15).
Results
The mRNA expression levels of JAK2 and PTPRC in PBMCs from patients with SLE and control subjects
One hundred thirty-five SLE patients were enrolled in this study. As shown in Table 1, clinical features of patients with SLE were grouped by disease activity. Based on quality control, a total of 120 patients with SLE (mean ± SD, 112 women, 36.50 ± 12.68 years; 8 men, 30.50 ± 15.42 years) and 129 control subjects (mean ± SD, women only, 36.09 ± 9.33 years) were included in the JAK2 gene expression statistic, and 122 SLE patients (mean ± SD, 114 women, 37.11 ± 12.69 years; 8 men, 30.50 ± 15.42 years) and 128 healthy control subjects (mean ± SD, women only, 36.12 ± 9.36 years) were included in the PTPRC gene expression statistic. As shown in Figs. 1 and 2, the median level of JAK2 mRNA expression in SLE patients was higher than that in controls (P < 0.05), and the median level of PTPRC mRNA expression in SLE patients was lower than that in healthy controls (P < 0.05).

mRNA expression levels of JAK2 were increased in SLE patients (n = 120) compared with those in healthy control subjects (n = 129; P = 0.005)

mRNA expression levels of PTPRC were increased in SLE patients (n = 122) compared with those in healthy control subjects (n = 128; P < 0.001)
Correlation of the rs1887428 variant with JAK2 mRNA expression levels in PBMCs
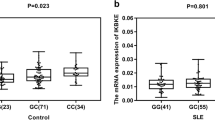
We conducted expression quantitative trait loci (eQTL) analysis to identify whether the rs1887428 SNP was associated with JAK2 mRNA expression levels in 129 healthy controls. However, there was no significant association observed between the rs1887428 SNP variant and JAK2 mRNA expression levels (P = 0.337) (Fig. 3).

Correlation of the SNP rs1887428 with JAK2 mRNA expression levels in PBMCs in healthy control subjects. Of the 129 controls, 66 individuals with CC, 46 with GC, and 17 with GG were analyzed. JAK2 mRNA expression did not significantly correlate with the rs1887428 genotype (P = 0.337)
Correlation of SLEDAI and clinical manifestations with JAK2 and PTPRC mRNA expression levels in PBMCs
To evaluate the relationship between two genes mRNA expression levels and SLEDAI, we respectively compared the two genes mRNA expression levels with active SLE (SLEDAI score ≥ 10) and inactive SLE (SLEDAI score < 10). As shown in Figs. 4 and 5, no statistical significance showed the SLEDAI score would affect JAK2 and PTPRC mRNA expression (P = 0.277 and 0.614, respectively). In addition, we found that PTPRC mRNA expression was significant with hematologic and arthritis involvement (P = 0.001 and 0.019, respectively) before the Bonferroni-corrected criteria. However, the association between PTPRC mRNA expression and hematologic involvement did not meet the Bonferroni-corrected significant criteria (P < 0.05/15) (Supplementary Tables 1 and 2).

No significant difference was found between the active SLE group and the inactive SLE group of JAK2 mRNA expression levels (P = 0.277)

No significant difference was found between the active SLE group and the inactive SLE group of PTPRC mRNA expression levels (P = 0.614)
Discussion
SLE is a chronic autoimmune disease associated with autoantibody production, inflammatory T cell activation, and aberrant cytokine and chemokine production. In this study, RNA samples were separated from PBMCs, which comprise T lymphocytes, B lymphocytes, and monocytes. A characteristic of SLE is the production of autoantibodies by autoreactive B lymphocytes. B cells can directly recognize antigens and secrete antibodies, which reinforce the adaptive immune response. Meanwhile, activated T cells can produce a variety of cytokines, including IL-2 and IL-17, which participate in signal transduction pathways associated with SLE pathogenesis [11,12,13]. The precursor of macrophages is monocytes, which can phagocytose and present antigens, secrete chemokines [14]. Moreover, monocyte contributes to renal damage and atherosclerosis in SLE [12].
Our previous study, which combined three GWASs comprising Chinese and European cohorts, discovered ten novel loci that were significantly associated with SLE: CD45, IKBKE, LBH, LPP-TPRG1-AS1, ATXN1, BACH2, GTF2I, RNASEH2C, ZFP90, and JAK2 [4]. However, the correlation between these genes and the occurrence and development of SLE is not explicit.
To explore the different mRNA expression levels of JAK2, we conducted qRT-PCR in patients with SLE and control subjects. The results showed that the mRNA expression levels of JAK2 in SLE patients were higher than those in control subjects. This result indicates that the increased levels of JAK2 mRNA expression may be related to the pathogenesis of SLE. The JAK2 gene encodes the JAK2 protein, which is a type of tyrosine kinase [15]. JAK2 is a member of the Janus kinase family, which comprises nonreceptor tyrosine kinases, and acts as a mainstay of the JAK/STAT signaling pathway, a cytokine receptor signaling pathway [16]. The JAK/STAT signaling pathway plays a critical role in the immune system by helping to fight pathogens and tumor cells and, more importantly, is significantly involved in autoimmune diseases. The activation of JAK/STAT signaling is triggered by a variety of inflammatory cytokines, including interleukins, type I (alpha/beta) interferons, type II (gamma) interferons, and so on [17]. According to the canonical JAK/STAT signaling pathway map, we found that some cytokines, including IL-6, IL-12 and IFN-γ, are particularly correlated with JAK2. The expression levels of these cytokines are significantly increased in SLE patients compared with the levels in healthy controls, similar to the JAK2 expression pattern. These cytokines participate in JAK/STAT signaling by activating the type I interferon signaling pathway [18,19,20]. Meanwhile, IFN-γ has been implicated as a key member of the IFN signaling pathway in SLE, and the IFN signaling pathway is upstream of the JAK/STAT signaling pathway [21]. In our study, we also found decreased levels of PTPRC mRNA expression in SLE patients. PTPRC, also known as CD45, encodes protein tyrosine phosphatase receptor C, which is a member of the PTP family. A previous study revealed that PTPRC suppresses JAK kinases and that damaged CD45 leads to the activation of JAKs and STAT proteins [5]. According to the aforementioned arguments, we can summarize that the decreased PTPRC expression may result in increased expression of JAK kinases, which likely leads to the development of SLE via JAK/STAT signaling.
In addition, our results indicated no association between JAK2 mRNA expression and the SNP rs1887428, which may be due to the limited sample size in this study. However, we can observe from Fig. 3 that the more instances of allele G in JAK2 were, the higher expression levels of JAK2 mRNA, indicating that rs1887428 may affect JAK2 mRNA expression. There were some limitations in our study, one of which was its cross-sectional nature involving patients with either advanced or stable disease. In addition, the limited sample size and use of corticosteroids or immunosuppressive medications for most SLE patients may lead to biases. Therefore, it is necessary for us to conduct further rigorous studies in a large sample size.
In conclusion, we revealed that JAK2 and PTPRC may play suggestive roles in the pathogenesis of SLE through JAK/STAT signaling. Further study is necessary to detect the true role of the JAK2 and PTPRC genes in SLE.
References
Osio-Salido E, Manapat-Reyes H (2010) Epidemiology of systemic lupus erythematosus in Asia. Lupus 19(12):1365–1373. https://doi.org/10.1177/0961203310374305
Wang J, Yang S, Chen JJ, Zhou SM, He SM, Liang YH, Meng W, Yan XF, Liu JJ, Ye DQ, Zhang XJ (2006) Systemic lupus erythematosus: a genetic epidemiology study of 695 patients from China. Arch Dermatol Res 298(10):485–491. https://doi.org/10.1007/s00403-006-0719-4
Deng Y, Tsao BP (2017) Updates in lupus genetics. Curr Rheumatol Rep 19(11):68. https://doi.org/10.1007/s11926-017-0695-z
Morris DL, Sheng Y, Zhang Y, Wang YF, Zhu Z, Tombleson P, Chen L, Cunninghame Graham DS, Bentham J, Roberts AL, Chen R, Zuo X, Wang T, Wen L, Yang C, Liu L, Yang L, Li F, Huang Y, Yin X, Yang S, Ronnblom L, Furnrohr BG, Voll RE, Schett G, Costedoat-Chalumeau N, Gaffney PM, Lau YL, Zhang X, Yang W, Cui Y, Vyse TJ (2016) Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat Genet 48(8):940–946. https://doi.org/10.1038/ng.3603
Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, Griffiths E, Krawczyk C, Richardson CD, Aitken K, Iscove N, Koretzky G, Johnson P, Liu P, Rothstein DM, Penninger JM (2001) CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 409(6818):349–354. https://doi.org/10.1038/35053086
Ghoreschi K, Laurence A, O’Shea JJ (2009) Janus kinases in immune cell signaling. Immunol Rev 228(1):273–287. https://doi.org/10.1111/j.1600-065X.2008.00754.x
Li J, Zhao S, Yi M, Hu X, Xie H, Zhu W, Chen M (2011) Activation of JAK-STAT1 signal transduction pathway in lesional skin and monocytes from patients with systemic lupus erythematosus. Zhong Nan Da Xue Xue Bao Yi Xue Ban 36(2):109–115. https://doi.org/10.3969/j.issn.1672-7347.2011.02.003
Hochberg MC (1997) Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40(9):1725. https://doi.org/10.1002/1529-0131(199709)40:9<1725::aid-art29>3.0.co;2-y
Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH (1992) Derivation of the SLEDAI. A disease activity index for lupus patients. The committee on prognosis studies in SLE. Arthritis Rheum 35(6):630–640
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Ohl K, Tenbrock K (2015) Regulatory T cells in systemic lupus erythematosus. Eur J Immunol 45(2):344–355. https://doi.org/10.1002/eji.201344280
Tsokos GC, Lo MS, Costa Reis P, Sullivan KE (2016) New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol 12(12):716–730. https://doi.org/10.1038/nrrheum.2016.186
Konya C, Paz Z, Tsokos GC (2014) The role of T cells in systemic lupus erythematosus: an update. Curr Opin Rheumatol 26(5):493–501. https://doi.org/10.1097/bor.0000000000000082
Chiu S, Bharat A (2016) Role of monocytes and macrophages in regulating immune response following lung transplantation. Curr Opin Organ Transplant 21(3):239–245. https://doi.org/10.1097/mot.0000000000000313
Duhe RJ, Rui H, Greenwood JD, Garvey K, Farrar WL (1995) Cloning of the gene encoding rat JAK2, a protein tyrosine kinase. Gene 158(2):281–285
Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G, Ihle JN (1998) Jak2 is essential for signaling through a variety of cytokine receptors. Cell 93(3):385–395
Cornez I, Yajnanarayana SP, Wolf AM, Wolf D (2017) JAK/STAT disruption induces immuno-deficiency: rationale for the development of JAK inhibitors as immunosuppressive drugs. Mol Cell Endocrinol 451:88–96. https://doi.org/10.1016/j.mce.2017.01.035
Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100(5):2610–2615. https://doi.org/10.1073/pnas.0337679100
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V (2003) Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197(6):711–723. https://doi.org/10.1084/jem.20021553
Bauer JW, Baechler EC, Petri M, Batliwalla FM, Crawford D, Ortmann WA, Espe KJ, Li W, Patel DD, Gregersen PK, Behrens TW (2006) Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med 3(12):e491. https://doi.org/10.1371/journal.pmed.0030491
Kawasaki M, Fujishiro M, Yamaguchi A, Nozawa K, Kaneko H, Takasaki Y, Takamori K, Ogawa H, Sekigawa I (2011) Possible role of the JAK/STAT pathways in the regulation of T cell-interferon related genes in systemic lupus erythematosus. Lupus 20(12):1231–1239. https://doi.org/10.1177/0961203311409963
Acknowledgments
We thank all the study participants and all the volunteers who have so willingly participated in this study, thus making this study possible.
Funding
This study was supported financially by the National Key Research and Development Program of China (2017YFC0909001 and 2016YFC0906102), the National Natural Science Foundation of China (81872527, 81803117, and 8132018016) and the Natural Science Foundation of Anhui Province (1808085QH284).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosures
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 22.1 kb)
Rights and permissions
About this article

Cite this article
Qian, D., Liu, L., Zhu, T. et al. JAK2 and PTPRC mRNA expression in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin Rheumatol 39, 443–448 (2020). https://doi.org/10.1007/s10067-019-04778-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-019-04778-w