
Abstract
Purpose of Review
Our understanding on genetic basis of SLE has been advanced through genome-wide association studies. We review recent progress in lupus genetics with a focus on SLE-associated loci that have been functionally characterized, and discuss the potential for clinical translation of genetics data.
Recent Findings
Over 100 loci have been confirmed to show robust association with SLE and many share with other immune-mediated diseases. Although causative variants captured at these established loci are limited, they guide biological studies of gene targets for functional characterization which highlight the importance of aberrant recognition of self-nucleic acid, type I interferon overproduction, and defective immune cell signaling underlying the pathogenesis of SLE. Increasing examples illustrate a predictive value of genetic findings in susceptibility/prognosis prediction, clinical classification, and pharmacological implication.
Summary
Genetic findings provide a foundation for better understanding of disease pathogenic mechanisms and opportunities for target selection in lupus drug development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
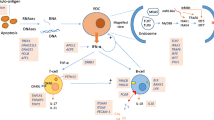
Systemic lupus erythematosus (SLE) is a prototypic autoimmune disease with a multifactorial etiology contributed by genetic, epigenetic, and environmental factors. Although the exact pathogenic mechanism of SLE remains elusive, impaired apoptosis/autophagy and clearance defects resulting in increased exposure of nuclear autoantigens, excessive activation of type I interferon (IFN-I) signaling via toll-like receptors (TLRs)/nucleic acid sensors, and altered immune cell signaling have long been recognized as key steps in the disease process [1]. Their importance has been strongly supported by genetics data, as defects in many pathway-related genes increase disease susceptibility or cause monogenic forms of SLE and/or SLE-like diseases (Fig. 1). In this review, we update extraordinary progress in SLE genetics, summarize the confirmed SLE susceptibility genes that are involved in major disease pathways, highlight loci associated with specific SLE features and shared among multiple immune-mediated diseases (IMDs), and discuss the potential for clinical translation of genetic findings.

Established SLE risk genes at each pathway. Asterisk: The SLE susceptibility genes with common variants are identified through GWAS, meta-analysis, fine-mapping, or replication studies yielding p < 5 × 10−8 in at least one ancestry. Genes labeled in red show association with SLE in multiple ancestries, while genes in black show association unique to one ancestry. Octothorpe: Genes with deficiency or rare mutations are associated with monogenic forms of SLE and/or SLE-like disease. Superscript lowercase letter a: NCF1 and CYBB defects causing chronic granulomatous disease (CGD). Superscript lowercase letter b: ADAR, IFIH1, TREX1, RNASEH2C/2B/2A, and SAMHD1 defects causing Aicardi-Goutières syndrome (AGS). Superscript lowercase letter c: TMEM173 defects causing STING-associated vasculopathy with onset in infancy (SAVI). Superscript lowercase letter d: ACP5 defects causing immuno-osseous dysplasia spondyloenchondrodysplasia (SPENCD). Superscript lowercase letter e: FAS and FASLG defects causing autoimmune lymphoproliferative syndrome (ALPS). Superscript lowercase letter f: SHOC2 defects causing Noonan syndrome with loose anagen hair (NSLAH). Superscript lowercase letter g: RAG1/2 defects causing severe combined immunodeficiency (SCID). Superscript lowercase letter h: KRAS and PTPN11 defects causing Noonan syndrome (NS)
SLE Susceptibility Genes with Common Variants
In most cases, genetic susceptibility of SLE fits the common disease-common variant hypothesis, which predicts that risk variants are present at > 1% throughout general populations and each confers a small effect (odds ratio (OR) usually < 1.5) accounting for a fraction of the overall genetic risk. During the last decade, genome-wide association studies (GWAS) followed by large-scale replication and meta-analyses have accelerated the discovery of common single nucleotide polymorphisms (SNPs) across the human genome associated with SLE susceptibility, yielding > 100 robust disease loci exceeding the genome-wide significance level (p < 5 × 10−8) predominantly in European and Asian ancestries [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16], most of which have also been confirmed in African-American or Amerindian/Hispanic populations [17, 18] (Fig. 1). Following the initial GWAS, collective efforts from fine-mapping studies, gene expression/epigenetic analyses, and mouse models help localize causative SNPs/target gene(s) within specific loci and to elucidate their functional impacts; however, many association intervals need to be further characterized. While few of these SNPs lie in the coding exons changing the function of its encoded protein, most lie in the noncoding regions affecting genes’ expression through transcriptional/posttranscriptional or epigenetic modifications. Here, we emphasize the SLE susceptibility loci (p < 5 × 10−8) that have likely causative variants localized (Table 1) [3,4,5,6,7, 10–11, 15, 17,18,19,20, 21••, 22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42, 43••, 44] and discuss their functional consequences implicated in the disease pathogenic pathways.
Increased Self-Antigens/Immune Complexes
Impaired apoptosis/autophagy and defective clearance may increase exposure of nuclear autoantigens to the immune system and allow deposition of immune complexes (ICs), contributing to the initiation and perpetuation of autoimmune responses in SLE.
Autophagy is a lysosomal pathway for degradation of intracellular components, including macroautophagy (also known as canonical autophagy), microautophagy, chaperone-mediated autophagy, and a recently defined non-canonical form LC3-associated phagocytosis (LAP). Autophagy-related processes can regulate many immune responses including clearance of microbes, antigen presentation, lymphocyte development, and cytokine secretion [45]; if disrupted, may cause autoimmune diseases. Activation of autophagy was observed in young pre-disease NZB/W F1 and MRLlpr/lpr mice, suggesting a genetic predisposition to dysregulated autophagy in murine lupus [46, 47]. This hypothesis is supported by human genetics data that multiple genes are related to autophagy pathway showing robust association with SLE, including ATG5 [7, 10] encoding a key component required for both canonical autophagy and LAP; NCF1 [21••] and NCF2 [19, 20] encoding two distinct regulatory subunits of phagocytic NADPH oxidase-2 (NOX2) complex whose activity is essential to LAP; and HIP1, ATG16L2, CDKN1B, DRAM1, and CLEC16A [7, 12,13,14,15,16, 48] with functions in modulation of autophagy. The likely causative variants of NCF1 (Arg90His) and NCF2 (His389Gln) show impressive effect size (OR > 2.5) and both risk alleles are associated with reduced NOX2-derived reactive oxygen species (ROS) production which is required for LAP to inhibit autoinflammatory lupus-like response to dying cells [19, 21••, 49, 50•]. However, whether and how these autophagy-related SLE risk gene loci are responsible for the reported dysregulation in autophagy of SLE patients [46, 51] remain to be clarified. Of note, these genes also have diverse functions, and investigation of autophagic and non-autophagic mechanisms will broaden our understanding of their contribution to the development of SLE.
Both rare deficiency and common variants in multiple members involved in IC clearance contribute to SLE susceptibility and manifestations (reviewed in [52]). ITGAM encodes CD11b together with CD18 that form the complement receptor 3 with functions in phagocytosis of complement-coated particles and ICs as well as regulation of leukocyte apoptosis, adhesion, and migration via interaction with ligands, such as ICAM1 that also shows genetic association with SLE [53]. Among the three missense variants of ITGAM (Arg77His, Ala858Val, and Pro1146Ser) that were associated with SLE, the Arg77His showed the strongest association. The mutant CD11b encoded by these SLE risk missense variants has impaired phagocytosis function in monocytes, neutrophils, and macrophages, leading to altered IC clearance, deposition, and tissue damage [54]. The reduced CD11b activity also has been connected to the chronically increased inflammatory status in SLE patients, as demonstrated by a recent study that CD11b could suppress TLR/IFN-I dependent inflammation via an AKT/FOXO3/IRF3/7 pathway and SLE patients carrying the ITGAM missense variants had elevated IFN-I levels [55•].
TLR/IFN-I Signaling
Given that IFN-I mediates multiple effects on immune responses including promotion of monocytes and plasmacytoid dendritic cell differentiation, activation of autoreactive T/B cells, stimulation of autoantibody production, and induction of pro-inflammatory cytokines and chemokines, dysregulated IFN-I signaling is thought to be central to the pathogenesis of SLE. In support of this, more than half of the identified SLE susceptibility genes encode proteins with functions directly or indirectly linked to IFN-I production or responses.
Increased exposure of nucleic acids contained in ICs or necrotic debris to endosomal receptors (e.g., TLR7) or cytosolic sensors (e.g., IFIH1) might represent a major trigger of IFN-I production in SLE. Studies in lupus-prone mouse models have clearly demonstrated the important role of TLR7 overexpression in driving lupus disease progression [56, 57]. A functional SNP at 3′untranslated region (UTR) of TLR7 is associated with SLE in four major ancestries [28, 29]. The SLE risk allele confers decreased degradation of TLR7 transcripts via reduced binding to an epigenetic factor, microRNA-3148, resulting in elevated TLR7 levels and heightened downstream IFN response [29]. IFIH1 encodes melanoma differentiation-associated protein 5 (MDA5), a member of RIG-I-like receptor family involved in the recognition of double-stranded RNA. Three common SNPs (two at exons and one at intron) are responsible for the genetic association of IFIH1 with SLE in multiple ancestries [24]. The SLE risk allele of the intronic SNP leads to decreased IFIH1 transcript levels by disrupting binding to protein complexes containing nucleolin and lupus autoantigen Ku70/80, which would be expected to promote autoantibody generation [24]. The SLE risk alleles of both missense SNPs confer phenotypic changes in apoptosis and inflammation-related gene expression [24].
Transcription factors downstream of endosomal TLRs, including IRF5, IRF7, and IRF8, control essentially IFN-I induction at the gene transcriptional level. Genetic variants in or near these three genes are associated with SLE across multiple ancestries, and likely causative variants at IRF5 and IRF7 loci have been localized. In addition to the known functional IRF5 polymorphisms [58], two independent genetic effects have been identified using computational approaches to model IRF5 association: one at the IRF5 promoter present in multiple ancestries and one in an 86-kb haplotype containing genes IRF5 and transportin 3 (TNPO3) present only in Europeans [26]. The SLE risk allele tagging the promoter effect correlates with elevated IRF5 expression through preferentially binding to a transcription factor ZBTB3 [26]. Integration of GWAS and expression quantitative-trait locus (eQTL) data leads to identification of a SNP located within the SLE-associated IRF7 haplotype that confers not only cis-eQTL effect on IRF7 expression but also trans-eQTL effect on regulating IFN responses in activated dendritic cells [27]. The X-linked gene IRAK1 encodes a kinase interacting with MyD88 required for nuclear factor-κB (NFκB) activation and TLR7/9-dependent induction of IFN-I. The missense variant of IRAK1 (Ser196Phe) shown to increase NFκB activity in vitro could capture a SLE risk haplotype containing nearby genes TMEM187 and MECP2 shared by multiple ancestries [30].
MicroRNA-146a (MIR146A) acts as a negative regulator to prevent overactivation of inflammatory responses by inhibiting key components involved in multiple immunological pathways including IFN-I pathway. Decreased mature miR-146a levels have been observed in SLE patients correlating with disease activity and upregulated expression of IFN response genes [59]. The SLE risk allele of a SNP at MIR146A gene promoter confers decreased miR-146a expression via its weak binding to transcription factor Ets-1 [25]. Of interest, IFN-I could inhibit miR-146a maturation through upregulation of monocyte chemotactic protein-induced protein 1, suggesting a non-genetic factor contributes to miR-146a dysregulation in SLE [60].
Nuclear Factor-κB Pathway
SLE susceptibility genes, including TNFAIP3 [4, 10, 31, 33], TNIP1 [6, 15], UBE2L3 [7, 15, 34, 35], PRKCB [48], and NFKBIA [61], encode proteins playing a regulatory role in the NFκB pathway. TNFAIP3 encodes an enzyme (A20) with both ubiquitin ligase and deubiquitinase activities to inhibit NFκB activation, TNF-mediated apoptosis, and NLRP3 inflammasome [62]. Studies in knockout mice have showed that defects in A20 expression or function predispose to autoimmunity by various mechanisms [63]. A pair of tandem polymorphic dinucleotides (TT>A) in a conserved regulatory region downstream of TNFAIP3 explains genetic association of TNFAIP3 with SLE [31]. The risk alleles with inefficient delivery of nuclear protein complex to the TNFAIP3 promoter via chromosomal looping attenuates A20 expression, leading to enhanced NFκB signaling activity in SLE [32]. UBE2L3, an E2 ubiquitin-conjugating enzyme, is required for the linear ubiquitin chain assembly complex-mediated activation of NFκB. A likely causal SNP at UBE2L3 promoter region tags a risk haplotype strongly associated with SLE [64]. The risk allele is correlated with increased UBE2L3 expression, enhanced NFκB activation, and increased circulating plasmablast/plasma cell numbers in SLE patients, supporting an importance of UBE2L3-regulated NFκB activation for B cell proliferation and differentiation [64].
Immune Cell Signaling
GWAS data has shown to contain dozens of SLE susceptibility genes that encode adaptor molecules, kinases, and cytokines participating in the regulation of immune cell activation, proliferation, and interaction (Fig. 1). Alterations in amount, structure, or function of these gene products perturbed by the identified SLE risk variants may lead to a loss of immune cell tolerance and sustained autoantibody production.
The human HLA region contains the class I and II regions that encode HLA molecules involved in antigen presentation, and the class III region that includes many genes encoding immune-related proteins (e.g., early complement components C2, C4, and factor B; cytokines TNF-α). Genetic association with SLE at the HLA region exhibits complex and multi-locus effects [65]. The highly conserved and extended haplotypes bearing the class II HLA-DR2 and HLA-DR3 alleles have long been recognized; however, the tight linkage disequilibrium (LD) across this locus has impeded dissecting the underlying causative variants. Using targeted sequencing combined with improved functional annotation analysis, a recent study indicated that all of the SLE-associated DR and DQ alleles are in strong LD with regulatory haplotypes termed as XL9, which could increase HLA class II gene transcription in a cis-specific fashion through interaction with regulatory elements such as CTCF [66•]. Increased surface expression of HLA class II molecules in immune cell lineages could impact a variety of functional processes leading to a hyperactive immune response commonly observed in SLE patients. The implication of class III genes in SLE susceptibility has been strongly supported by GWAS and fine-mapping studies [5, 7, 15, 67], such as MSH5 with function in DNA repair; and RDBP, SKIV2L, DOM3Z, and STK19 involved in RNA processing, but the causative alleles remain to be identified requiring more powerful statistical methods to better decipher the extensive LD structure.
The lymphoid-specific tyrosine phosphatase (LYP, encoded by PTPN22) physically interacts with c-Src tyrosine kinase (encoded by CSK) to regulate signal transduction in lymphocytes. Both genes have well-known variants associated with multiple autoimmune diseases including SLE. The SLE risk allele at a CSK intronic polymorphism is associated with elevated CSK expression, increased downstream Src kinase (Lyn) phosphorylation, enhanced BCR-mediated mature B cell activation and transitional B cell expansion, supporting a role for CSK involved in multiple B cell developmental stages [44]. The SLE-associated Arg620Trp of PTPN22 is a gain-of-function variant encoding a more active phosphatase. Humans carrying the risk 620Trp allele and knockin mice expressing the syntenic 619Trp mutation show altered T cell receptor (TCR) and BCR signaling with enhanced B cell autoreactivity [68]. In naive and effector T cells, PTPN22 can limit TCR signaling by weak agonists and self-antigens but not impeding responses to strong agonist antigens, which may explain the population expansion of effector and memory T cells observed in humans and mice with either PTPN22 variants or Ptpn22 −/− alleles [69]. In addition to lymphocytes, the risk 620Trp allele is associated with diminished TLR-induced IFN-I production in myeloid cells [70], but with enhanced functions in neutrophils including increased transendothelial migration, Ca2+ release, and ROS production [71].
BLK encodes a member of the Src family kinases that typically functions in intracellular signaling and regulation of B cell proliferation, differentiation, and tolerance. The SLE risk variants localized at the promoter of BLK confer reduced promoter activity in B cell lines representing different B cell developmental stages, suggesting that decreased BLK expression may affect development and functional responses in B cells [41]. BANK1 encodes an adaptor/scaffold protein that facilitates intracellular calcium release and alters B cell activation threshold. The study in Bank1-deficient B6.Sle1.yaa lupus mouse model shows that Bank1 controls Tlr7-induced signaling in B cells to regulate IgG production, supporting BANK1 as a SLE susceptibility gene [72]. Three functional variants including one intronic and two missense SNPs of BANK1 are associated with SLE, which contributes to lupus by decreasing B cell signaling and enhancing expansion of memory B cells [3, 73]. PXK encodes a phox domain-containing protein, which binds to and modulates Na, K-ATPase subunits involved in the regulation of synaptic transmission. Knockdown of PXK leads to reduced BCR internalization, implicating a direct involvement of PXK in BCR trafficking [40]. Genetic association of the PXK region with SLE has been identified only in European population, which could be tagged by a haplotype containing highly linked variants in the promoter and first exon of PXK [5, 7, 40]. Individuals carrying the SLE risk haplotype exhibit a decreased rate of BCR internalization, suggesting a mechanism model in which PXK contributes to lupus through regulating B cell survival and cell fate [40].
Transcription factors encoded by the SLE susceptibility genes, including ETS1, IKZF1, IKZF2, and IKZF3, play an important role in regulating immune cell signaling. Maintaining appropriate ETS1 levels is essential to prevent loss of B cell tolerance [74]. Ets1-deficient mice develop a lupus-like disease characterized by high titers of autoantibodies and IC deposition in the kidney [75]. Decreased ETS1 mRNA levels were reported in peripheral blood mononuclear cells (PBMCs) of SLE patients [11]. A trans-ancestral fine-mapping study localizes the SLE-associated signal to a downstream variant that could lead increased binding of an activated transcription factor pSTAT1 and is associated with reduced ETS1 expression [42]. IKZF1, IKZF2, and IKZF3 encode three members of the Ikaros family of zinc finger proteins. Their genetic associations with SLE have been identified in European and Asian populations [7, 15, 76]; however, the likely causative variants and the functional mechanism remain elusive. A trans-eQTL study revealed that the SLE risk IKZF1 variant not only alters IKZF1 levels in cis but also regulates expression of C1QB and five IFN-I response genes in trans, demonstrating that trans-eQTL mapping could reveal downstream effects of the disease-associated variants [77•].
IL-10 is an immunoregulatory cytokine ubiquitously expressed in immune cells. The observation of elevated serum IL-10 levels in SLE patients correlating with disease activity and promising findings of anti-IL-10 monoclonal antibody treatment in SLE patients support a rationale to understand how IL-10 production is regulated. From a genetic study, the SLE risk allele of a regulatory polymorphism at IL10 upstream confers increased IL10 expression by preferentially binding to an activated transcription factor pElk-1 associated with disease activity in SLE B cells [39]. An epigenetic mechanism also involved that reduced DNA methylation and trans-activation of IL10 transcription by increased Stat3/Stat5 recruitment to the IL10 regulatory regions could promote IL10 expression in SLE T cells [78]. B-cell activating factor (BAFF) is a cytokine in both membrane-bound and soluble forms mainly produced by monocytes and neutrophils, and acts as a homeostatic regulator for B cell selection and survival. Overexpression of BAFF in lupus mouse models leads to autoimmunity and BAFF antagonism conversely improves self-tolerance. Neutralizing BAFF by the FDA-approved drug belimumab (anti-BAFF monoclonal antibody) has demonstrated clinical efficacy in SLE patients. A GWAS in Sardinians identified an insertion-deletion variant, GCTGT → A (referred to as BAFF-var), at 3′UTR of TNFSF13B (encoding BAFF) as a likely causal variant associated with SLE and multiple sclerosis [43••]. This BAFF-var variant creates an alternative polyadenylation motif, AATAAA, from the sequence AAT[GCTGT/A]AA which yields a shorter transcript less inhibited by microRNA-15a, resulting in increased soluble BAFF production and upregulated humoral immunity [43••]. Population genetic signatures suggested that the high frequency of this variant in Sardinians might evolved by positive selection due to its enhanced resistance to malaria [43••]. As SLE patients with higher basal levels of BAFF show poorer clinical responses to B cell depletion therapy (rituximab), patients carrying BAFF-var might have a differential benefit from BAFF-directed therapies and a weaker response to rituximab.
Other Loci
GWAS have discovered an expanding number of novel loci showing robust association with SLE. Few loci encode gene products with immune functions relevant to but not fully elucidated in the disease pathogenesis (such as TET3 involved in DNA demethylation), most encode gene products with unknown immune functions requiring further characterization. To understand how they generate disease risk may reveal new pathogenic mechanisms for SLE.
Copy Number Variations
In addition to SNPs, another genetic diversity associated with complex diseases is copy number variations (CNVs) that generally include deletion, insertion, and duplication of genomic regions disrupting gene dosage and/or structure. Studies of C4, FCGR3B/FCGR2C, TLR7, CFHR3/CFHR1, and NCF1 have highlighted a role of gene CNVs in SLE [21••, 79, 80]. For example, the deletion of CFHR3/CFHR1 which might dysregulate complement activation showed dosage-dependent association with SLE in multiple populations [80]. NCF1 is located within large DNA duplications at 7q11.23 and has 98% sequence identity with the nonfunctional pseudogenes NCF1B and NCF1C. The GTGT sequence in the second exon of NCF1 is a well-characterized variant that can distinguish NCF1 from NCF1B/NCF1C which contain a GT deletion (ΔGT). The GTGT to ΔGT mutation in NCF1 causes a premature stop codon leading to absence of functional NCF1 protein, while ΔGT to GTGT mutation in NCF1B/NCF1C are thought to produce intact NCF1 protein. Decreased (0 and 1 copy) and increased (≥ 3 copies) CNVs of NCF1 predispose to and protect against SLE, respectively [21••]. More CNVs are being identified through various advanced genome-wide technologies, and integration of CNVs with SNPs will improve our understanding of the disease susceptibility.
Gene Defects in Monogenic Forms of SLE and SLE-Like Disease
Few cases of SLE, especially early-onset juvenile SLE, are associated with rare but highly penetrant single-gene mutations, such as a complete deficiency in one of the classical complement pathway genes (C1Q, C1R/S, C2, C4A, and C4B) and a nonsense mutation in deoxyribonuclease I (encoded by DNASE1) [81, 82]. Some monogenic disorders with lupus-like features have been reported; although rare, these extreme phenotypes help to identify disease causation. For example, studies of the Aicardi-Goutières syndrome and the related familial chilblain lupus have identified mutations in genes involved in cytosolic nucleic acid sensing and IFN-I production pathway, including TREX1, SAMHD1, ADAR, IFIH1, and RNASEH2A/2B/2C [83]. A newly described autoinflammatory disease termed as infantile-onset STING-associated vasculopathy is also caused by gain-of-function mutations of an IFN-I pathway gene TMEM173 [84]. Additional examples include genes in apoptosis/autophagy pathway (e.g., PRKCD) and lymphocyte signaling (reviewed in [1, 85]). Characterization of gene defects in monogenic forms of SLE and SLE-like disease highlights the importance of aberrant recognition of self-nucleic acid and IFN-I overproduction underlying the pathogenesis of SLE.
Genes Shared Among IMDs
In addition to SLE, rapid progress has also been made in GWAS of other IMDs, including rheumatoid arthritis (RA), juvenile idiopathic arthritis (JIA), ankylosing spondylitis (AS), systemic sclerosis (SSc), Sjögren’s syndrome (SS), psoriasis (PSO), type 1 diabetes (T1D), autoimmune thyroid disease (AITD), inflammatory bowel disease (IBD, including Crohn’s disease and ulcerative colitis), celiac disease (CEL), multiple sclerosis (MS), and primary biliary cirrhosis (PBC). The availability of “ImmunoChip” microarray, which interrogates ∼ 200,000 polymorphisms covering 186 loci with established disease association, has accelerated immunogenetics gene mapping [86]. As expected, many SLE susceptibility loci overlapped across all of other IMDs (Table 2). However, sharing is complex as the most associated variant(s) at a shared locus often differs, such as different haplotypes of HLA class II alleles with IMDs. Even if a shared locus harbors the same variant, it may confer opposite associations (e.g., PTPN22 620Trp allele increases risk for SLE but is protective for IBD) or different effect sizes among diseases of the same population (e.g., NCF1 90His allele confers strong, modest, and weak effect size for SLE, SS, and RA, respectively). Identification of genes/loci shared by SLE with other diseases supports the existence of overlapping etiological mechanisms among IMDs and will expand our understanding of increased risk in developing SLE concomitant with multiple IMDs.
Clinical Translation of Genetic Findings
Emerging examples demonstrate the potential for GWAS findings could lead to disease risk prediction, clinical classification, and drug development [87]. Compared to a single locus that often confers only a small disease risk, calculation of genetic risk score (GRS) by counting the number of SLE-associated risk alleles weighted by ORs has more power to predict disease susceptibility and/or progression, which revealed a higher cumulative genetic load in (i) non-European (especially African) than European individuals, which may help explain the increased prevalence of SLE in non-Europeans [8]; (ii) men than women SLE patients of European descent [88]; and (iii) childhood-onset than adult-onset SLE patients of African ancestry [89]. Of note, the established SLE-associated SNPs used in GRS calculation explain ~ 28% of the disease heritability [16] so that such comparison could not entirely reflect genetic risk for SLE among populations. It is anticipated that the GRS predictive value will increase upon more SLE risk variants, particularly those with high effect size (OR > 2), continue to be identified.
The clinically heterogeneous presentation of SLE has inspired genotype-phenotype association analyses, which confirmed a common genetic contribution to disease susceptibility and specific subphenotypes [90]. ITGAM, an important member involved in IC clearance process, represents an example showing genetic association not only with SLE susceptibility, but also with skin, renal, joint, and neurological disorders as well as anti-dsDNA autoantibody [91,92,93,94]. Genetic factors contribute to disease susceptibility and subphenotypes/prognosis might not overlap as shown for patients with lupus nephritis (LN) or end-stage renal disease (ESRD) in whom non-SLE susceptibility loci for LN or ESRD have been identified, such as PDGRFA, ID4, HAS2, SLC5A11, MYH9, and APOL1 [95, 96]. This is also demonstrated in Crohn’s patients that none of the prognosis-associated loci showed significant association with disease susceptibility [97]. Reanalysis of existing SLE GWAS data using selected subphenotypes may yield new risk loci for organ involvement or disease progression.
Attempt to implement genetic findings for lupus drug development is underway, which requires mapping SLE-associated variants to genes encoding druggable proteins and/or drug-like compounds, similarly as a study in RA [98•]. Efforts to expand the set of genes comprising the druggable genome will accelerate drug target identification and validation in lupus [99•].
Conclusions
GWAS and post-GWAS studies have mapped an extraordinary growing number of genes/loci showing convincing association with SLE, building a solid foundation for better understanding of disease pathogenic mechanisms and target selection in drug development. Future efforts prioritize the identification of causal and functional variants within many association intervals as well as new associations including rare variants to explain the missing heritability of SLE. Environmental risk factors may interact with genetic factors specifically to affect disease development as supported by a recent study showing a combined role for vitamin D status and vitamin D gene variants in the transition to SLE [100]. As increasing knowledge has being gained from genomic, epigenomic, and biological studies, improved preventive and therapeutic strategy for lupus patient management could be expected.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Tsokos GC, Lo MS, Reis PC, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12(12):716–30. https://doi.org/10.1038/nrrheum.2016.186.
Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008;358(9):900–9. https://doi.org/10.1056/NEJMoa0707865.
Kozyrev SV, Abelson AK, Wojcik J, Zaghlool A, Linga Reddy MV, Sanchez E, et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet. 2008;40(2):211–6. https://doi.org/10.1038/ng.79.
Graham RR, Cotsapas C, Davies L, Hackett R, Lessard CJ, Leon JM, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40(9):1059–61. https://doi.org/10.1038/ng.200.
Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40(2):204–10. https://doi.org/10.1038/ng.81.
Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41(11):1228–33. https://doi.org/10.1038/ng.468.
Bentham J, Morris DL, Cunninghame Graham DS, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47(12):1457–64. https://doi.org/10.1038/ng.3434.
Morris DL, Sheng Y, Zhang Y, Wang YF, Zhu Z, Tombleson P, et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat Genet. 2016;48(8):940–6. https://doi.org/10.1038/ng.3603.
Langefeld CD, Ainsworth HC, Cunninghame Graham DS, Kelly JA, Comeau ME, Marion MC, et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat Commun. 2017;8:16021. https://doi.org/10.1038/ncomms16021.
Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41(11):1234–7. https://doi.org/10.1038/ng.472.
Yang W, Shen N, Ye DQ, Liu Q, Zhang Y, Qian XX, et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010;6(2):e1000841. https://doi.org/10.1371/journal.pgen.1000841.
Okada Y, Shimane K, Kochi Y, Tahira T, Suzuki A, Higasa K, et al. A genome-wide association study identified AFF1 as a susceptibility locus for systemic lupus eyrthematosus in Japanese. PLoS Genet. 2012;8(1):e1002455. https://doi.org/10.1371/journal.pgen.1002455.
Yang W, Tang H, Zhang Y, Tang X, Zhang J, Sun L, et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am J Hum Genet. 2013;92(1):41–51. https://doi.org/10.1016/j.ajhg.2012.11.018.
Lessard CJ, Sajuthi S, Zhao J, Kim K, Ice JA, Li H, et al. Identification of a systemic lupus erythematosus risk locus spanning ATG16L2, FCHSD2, and P2RY2 in Koreans. Arthritis Rheumatol. 2016;68(5):1197–209. https://doi.org/10.1002/art.39548.
Sun C, Molineros JE, Looger LL, Zhou XJ, Kim K, Okada Y, et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet. 2016;48(3):323–30. https://doi.org/10.1038/ng.3496.
Molineros JE, Yang W, Zhou XJ, Sun C, Okada Y, Zhang H, et al. Confirmation of five novel susceptibility loci for systemic lupus erythematosus (SLE) and integrated network analysis of 82 SLE susceptibility loci. Hum Mol Genet. 2017;26(6):1205–16. https://doi.org/10.1093/hmg/ddx026.
Sanchez E, Comeau ME, Freedman BI, Kelly JA, Kaufman KM, Langefeld CD, et al. Identification of novel genetic susceptibility loci in African American lupus patients in a candidate gene association study. Arthritis Rheum. 2011;63(11):3493–501. https://doi.org/10.1002/art.30563.
Alarcon-Riquelme ME, Ziegler JT, Molineros J, Howard TD, Moreno-Estrada A, Sanchez-Rodriguez E, et al. Genome-wide association study in an Amerindian ancestry population reveals novel systemic lupus erythematosus risk loci and the role of European admixture. Arthritis Rheumatol. 2016;68(4):932–43. https://doi.org/10.1002/art.39504.
Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, et al. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci U S A. 2012;109(2):E59–67. https://doi.org/10.1073/pnas.1113251108.
Kim-Howard X, Sun C, Molineros JE, Maiti AK, Chandru H, Adler A, et al. Allelic heterogeneity in NCF2 associated with systemic lupus erythematosus (SLE) susceptibility across four ethnic populations. Hum Mol Genet. 2014;23(6):1656–68. https://doi.org/10.1093/hmg/ddt532.
•• Zhao J, Ma J, Deng Y, Kelly JA, Kim K, Bang SY, et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat Genet. 2017;49(3):433–7. https://doi.org/10.1038/ng.3782. Identification of NCF1 as the likely causal gene driving a strong SLE-associated signal detected by GWAS within the GTF2IRD1-GTF2I-NCF1 region at chromosome 7q11.23
Zhu XW, Wang Y, Wei YH, Zhao PP, Wang XB, Rong JJ, et al. Comprehensive assessment of the association between FCGRs polymorphisms and the risk of systemic lupus erythematosus: evidence from a meta-analysis. Sci Rep. 2016;6:31617. https://doi.org/10.1038/srep31617.
Lee YH, Bae SC. Association between the functional ITGAM rs1143679 G/A polymorphism and systemic lupus erythematosus/lupus nephritis or rheumatoid arthritis: an update meta-analysis. Rheumatol Int. 2015;35(5):815–23. https://doi.org/10.1007/s00296-014-3156-2.
Molineros JE, Maiti AK, Sun C, Looger LL, Han S, Kim-Howard X, et al. Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PLoS Genet. 2013;9(2):e1003222. https://doi.org/10.1371/journal.pgen.1003222.
Luo X, Yang W, Ye DQ, Cui H, Zhang Y, Hirankarn N, et al. A functional variant in microRNA-146a promoter modulates its expression and confers disease risk for systemic lupus erythematosus. PLoS Genet. 2011;7(6):e1002128. https://doi.org/10.1371/journal.pgen.1002128.
Kottyan LC, Zoller EE, Bene J, Lu X, Kelly JA, Rupert AM, et al. The IRF5-TNPO3 association with systemic lupus erythematosus has two components that other autoimmune disorders variably share. Hum Mol Genet. 2015;24(2):582–96. https://doi.org/10.1093/hmg/ddu455.
Lee MN, Ye C, Villani AC, Raj T, Li W, Eisenhaure TM, et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science. 2014;343(6175):1246980. https://doi.org/10.1126/science.1246980.
Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2010;107:15838–43. https://doi.org/10.1073/pnas.1001337107.
Deng Y, Zhao J, Sakurai D, Kaufman KM, Edberg JC, Kimberly RP, et al. MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS Genet. 2013;9(2):e1003336. https://doi.org/10.1371/journal.pgen.1003336.
Kaufman KM, Zhao J, Kelly JA, Hughes T, Adler A, Sanchez E, et al. Fine mapping of Xq28: both MECP2 and IRAK1 contribute to risk for systemic lupus erythematosus in multiple ancestral groups. Ann Rheum Dis. 2013;72(3):437–44. https://doi.org/10.1136/annrheumdis-2012-201851.
Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet. 2011;43(3):253–8. https://doi.org/10.1038/ng.766.
Wang S, Wen F, Wiley GB, Kinter MT, Gaffney PM. An enhancer element harboring variants associated with systemic lupus erythematosus engages the TNFAIP3 promoter to influence A20 expression. PLoS Genet. 2013;9(9):e1003750. https://doi.org/10.1371/journal.pgen.1003750.
Moaaz M, Mohannad N. Association of the polymorphisms of TRAF1 (rs10818488) and TNFAIP3 (rs2230926) with rheumatoid arthritis and systemic lupus erythematosus and their relationship to disease activity among Egyptian patients. Cent Eur J Immunol. 2016;41(2):165–75. https://doi.org/10.5114/ceji.2016.60991.
Agik S, Franek BS, Kumar AA, Kumabe M, Utset TO, Mikolaitis RA, et al. The autoimmune disease risk allele of UBE2L3 in African American patients with systemic lupus erythematosus: a recessive effect upon subphenotypes. J Rheumatol. 2012;39(1):73–8. https://doi.org/10.3899/jrheum.110590.
Wang S, Adrianto I, Wiley GB, Lessard CJ, Kelly JA, Adler AJ, et al. A functional haplotype of UBE2L3 confers risk for systemic lupus erythematosus. Genes Immun. 2012;13(5):380–7. https://doi.org/10.1038/gene.2012.6.
Namjou B, Kim-Howard X, Sun C, Adler A, Chung SA, Kaufman KM, et al. PTPN22 association in systemic lupus erythematosus (SLE) with respect to individual ancestry and clinical sub-phenotypes. PLoS One. 2013;8(8):e69404. https://doi.org/10.1371/journal.pone.0069404.
Tang L, Wang Y, Zheng S, Bao M, Zhang Q, Li J. PTPN22 polymorphisms, but not R620W, were associated with the genetic susceptibility of systemic lupus erythematosus and rheumatoid arthritis in a Chinese Han population. Hum Immunol. 2016;77(8):692–8. https://doi.org/10.1016/j.humimm.2016.04.021.
Elghzaly AA, Metwally SS, El-Chennawi FA, Elgayaar MA, Mosaad YM, El-Toraby EE, et al. IRF5, PTPN22, CD28, IL2RA, KIF5A, BLK and TNFAIP3 genes polymorphisms and lupus susceptibility in a cohort from the Egypt Delta; relation to other ethnic groups. Hum Immunol. 2015;76(7):525–31. https://doi.org/10.1016/j.humimm.2015.06.001.
Sakurai D, Zhao J, Deng Y, Kelly JA, Brown EE, Harley JB, et al. Preferential binding to Elk-1 by SLE-associated IL10 risk allele upregulates IL10 expression. PLoS Genet. 2013;9(10):e1003870. https://doi.org/10.1371/journal.pgen.1003870.
Vaughn SE, Foley C, Lu X, Patel ZH, Zoller EE, Magnusen AF, et al. Lupus risk variants in the PXK locus alter B-cell receptor internalization. Front Genet. 2015;5:450. https://doi.org/10.3389/fgene.2014.00450.
Guthridge JM, Lu R, Sun H, Sun C, Wiley GB, Dominguez N, et al. Two functional lupus-associated BLK promoter variants control cell-type- and developmental-stage-specific transcription. Am J Hum Genet. 2014;94(4):586–98. https://doi.org/10.1016/j.ajhg.2014.03.008.
Lu X, Zoller EE, Weirauch MT, Wu Z, Namjou B, Williams AH, et al. Lupus risk variant increases pSTAT1 binding and decreases ETS1 expression. Am J Hum Genet. 2015;96(5):731–9. https://doi.org/10.1016/j.ajhg.2015.03.002.
•• Steri M, Orru V, Idda ML, Pitzalis M, Pala M, Zara I, et al. Overexpression of the cytokine BAFF and autoimmunity risk. N Engl J Med. 2017;376(17):1615–26. https://doi.org/10.1056/NEJMoa1610528. Identification of a TNFSF13B variant associated with multiple sclerosis and SLE, and its effect at the population, cellular, and molecular levels
Manjarrez-Orduno N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, et al. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat Genet. 2012;44(11):1227–30. https://doi.org/10.1038/ng.2439.
Lapaquette P, Guzzo J, Bretillon L, Bringer MA. Cellular and molecular connections between autophagy and inflammation. Mediat Inflamm. 2015;2015:398483. https://doi.org/10.1155/2015/398483.
Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2015;74(5):912–20. https://doi.org/10.1136/annrheumdis-2013-204343.
Gros F, Arnold J, Page N, Decossas M, Korganow AS, Martin T, et al. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy. 2012;8(7):1113–23. https://doi.org/10.4161/auto.20275.
Demirci FY, Wang X, Kelly JA, Morris DL, Barmada MM, Feingold E, et al. Identification of a new susceptibility locus for systemic lupus erythematosus on chromosome 12 in individuals of European ancestry. Arthritis Rheumatol. 2016;68(1):174–83. https://doi.org/10.1002/art.39403.
Olsson LM, Johansson AC, Gullstrand B, Jonsen A, Saevarsdottir S, Ronnblom L, et al. A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann Rheum Dis. 2017;76(9):1607–13. https://doi.org/10.1136/annrheumdis-2017-211287.
• Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–9. https://doi.org/10.1038/nature17950. This study provides evidence for LC3-associated phagocytosis in the clearance of dying cells and inflammation in the control of SLE
Alessandri C, Barbati C, Vacirca D, Piscopo P, Confaloni A, Sanchez M, et al. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. FASEB J. 2012;26(11):4722–32. https://doi.org/10.1096/fj.12-206060.
Deng Y, Tsao BP. Genetics of human SLE. In: Wallace DJ, Hahn BH, editors. Dubois’ lupus erythematosus and related syndromes. Philadephia: Elsevier Inc; 2012. p. 35–45.
Kim K, Brown EE, Choi CB, Alarcon-Riquelme ME, Kelly JA, Glenn SB, et al. Variation in the ICAM1-ICAM4-ICAM5 locus is associated with systemic lupus erythematosus susceptibility in multiple ancestries. Ann Rheum Dis. 2012;71(11):1809–14. https://doi.org/10.1136/annrheumdis-2011-201110.
Fossati-Jimack L, Ling GS, Cortini A, Szajna M, Malik TH, McDonald JU, et al. Phagocytosis is the main CR3-mediated function affected by the lupus-associated variant of CD11b in human myeloid cells. PLoS One. 2013;8(2):e57082. https://doi.org/10.1371/journal.pone.0057082.
• Faridi MH, Khan SQ, Zhao W, Lee HW, Altintas MM, Zhang K, et al. CD11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J Clin Invest. 2017;127(4):1271–83. https://doi.org/10.1172/JCI88442. This study provides evidence for CD11b linked to TLR/IFN-I-dependent inflammation
Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27(5):801–10. https://doi.org/10.1016/j.immuni.2007.09.009.
Das A, Heesters BA, Bialas A, O'Flynn J, Rifkin IR, Ochando J, et al. Follicular dendritic cell activation by TLR ligands promotes autoreactive B cell responses. Immunity. 2017;46(1):106–19. https://doi.org/10.1016/j.immuni.2016.12.014.
Jensen MA, Niewold TB. Interferon regulatory factors: critical mediators of human lupus. Transl Res. 2015;165(2):283–95. https://doi.org/10.1016/j.trsl.2014.10.002.
Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60(4):1065–75. https://doi.org/10.1002/art.24436.
Qu B, Cao J, Zhang F, Cui H, Teng J, Li J, et al. Type I interferon inhibition of microRNA-146a maturation through up-regulation of monocyte chemotactic protein-induced protein 1 in systemic lupus erythematosus. Arthritis Rheumatol. 2015;67(12):3209–18. https://doi.org/10.1002/art.39398.
Li Y, Cheng H, Zuo XB, Sheng YJ, Zhou FS, Tang XF, et al. Association analyses identifying two common susceptibility loci shared by psoriasis and systemic lupus erythematosus in the Chinese Han population. J Med Genet. 2013;50(12):812–8. https://doi.org/10.1136/jmedgenet-2013-101787.
Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol. 2014;35(1):22–31. https://doi.org/10.1016/j.it.2013.10.005.
Vereecke L, Beyaert R, van Loo G. Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochem Soc Trans. 2011;39(4):1086–91. https://doi.org/10.1042/BST0391086.
Lewis MJ, Vyse S, Shields AM, Boeltz S, Gordon PA, Spector TD, et al. UBE2L3 polymorphism amplifies NF-kappaB activation and promotes plasma cell development, linking linear ubiquitination to multiple autoimmune diseases. Am J Hum Genet. 2015;96(2):221–34. https://doi.org/10.1016/j.ajhg.2014.12.024.
Morris DL, Taylor KE, Fernando MM, Nititham J, Alarcon-Riquelme ME, Barcellos LF, et al. Unraveling multiple MHC gene associations with systemic lupus erythematosus: model choice indicates a role for HLA alleles and non-HLA genes in Europeans. Am J Hum Genet. 2012;91(5):778–93. https://doi.org/10.1016/j.ajhg.2012.08.026.
• Raj P, Rai E, Song R, Khan S, Wakeland BE, Viswanathan K, et al. Regulatory polymorphisms modulate the expression of HLA class II molecules and promote autoimmunity. elife. 2016:5. https://doi.org/10.7554/eLife.12089. Target sequencing identified the SLE-associated HLA-DR and HLA - DQ alleles in strong LD with regulatory haplotypes termed as XL9, which could increase HLA class II genes transcription in a cis-specific fashion
Fernando MM, Freudenberg J, Lee A, Morris DL, Boteva L, Rhodes B, et al. Transancestral mapping of the MHC region in systemic lupus erythematosus identifies new independent and interacting loci at MSH5, HLA-DPB1 and HLA-G. Ann Rheum Dis. 2012;71(5):777–84. https://doi.org/10.1136/annrheumdis-2011-200808.
Bottini N, Peterson EJ. Tyrosine phosphatase PTPN22: multifunctional regulator of immune signaling, development, and disease. Annu Rev Immunol. 2014;32:83–119. https://doi.org/10.1146/annurev-immunol-032713-120249.
Salmond RJ, Brownlie RJ, Morrison VL, Zamoyska R. The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nat Immunol. 2014;15(9):875–83. https://doi.org/10.1038/ni.2958.
Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity. 2013;39(1):111–22. https://doi.org/10.1016/j.immuni.2013.06.013.
Bayley R, Kite KA, McGettrick HM, Smith JP, Kitas GD, Buckley CD, et al. The autoimmune-associated genetic variant PTPN22 R620W enhances neutrophil activation and function in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis. 2014;74(8):1588–95. https://doi.org/10.1136/annrheumdis-2013-204796.
Wu YY, Kumar R, Iida R, Bagavant H, Alarcon-Riquelme ME. BANK1 regulates IgG production in a lupus model by controlling TLR7-dependent STAT1 activation. PLoS One. 2016;11(5):e0156302. https://doi.org/10.1371/journal.pone.0156302.
Dam EM, Habib T, Chen J, Funk A, Glukhova V, Davis-Pickett M, et al. The BANK1 SLE-risk variants are associated with alterations in peripheral B cell signaling and development in humans. Clin Immunol. 2016;173:171–80. https://doi.org/10.1016/j.clim.2016.10.018.
Russell L, John S, Cullen J, Luo W, Shlomchik MJ, Garrett-Sinha LA. Requirement for transcription factor Ets1 in B cell tolerance to self-antigens. J Immunol. 2015;195(8):3574–83. https://doi.org/10.4049/jimmunol.1500776.
Wang D, John SA, Clements JL, Percy DH, Barton KP, Garrett-Sinha LA. Ets-1 deficiency leads to altered B cell differentiation, hyperresponsiveness to TLR9 and autoimmune disease. Int Immunol. 2005;17(9):1179–91. https://doi.org/10.1093/intimm/dxh295.
Cai X, Qiao Y, Diao C, Xu X, Chen Y, Du S, et al. Association between polymorphisms of the IKZF3 gene and systemic lupus erythematosus in a Chinese Han population. PLoS One. 2014;9(10):e108661. https://doi.org/10.1371/journal.pone.0108661.
• Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45(10):1238–43. https://doi.org/10.1038/ng.2756. This eQTL meta-analysis identifies trans -eQTLs for 233 SNPs that are associated with complex traits, yielding insights into downstream effects of disease-associated variants
Hedrich CM, Rauen T, Apostolidis SA, Grammatikos AP, Rodriguez Rodriguez N, Ioannidis C, et al. Stat3 promotes IL-10 expression in lupus T cells through trans-activation and chromatin remodeling. Proc Natl Acad Sci U S A. 2014;111(37):13457–62. https://doi.org/10.1073/pnas.1408023111.
Ptacek T, Li X, Kelley JM, Edberg JC. Copy number variants in genetic susceptibility and severity of systemic lupus erythematosus. Cytogenet Genome Res. 2008;123(1–4):142–7. https://doi.org/10.1159/000184701.
Zhao J, Wu H, Khosravi M, Cui H, Qian X, Kelly JA, et al. Association of genetic variants in complement factor h and factor h-related genes with systemic lupus erythematosus susceptibility. PLoS Genet. 2011;7(5):e1002079. https://doi.org/10.1371/journal.pgen.1002079.
Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity. 2007;40(8):560–6. https://doi.org/10.1080/08916930701510673.
Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. 2001;28(4):313–4. https://doi.org/10.1038/91070.
Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;167A(2):296–312. https://doi.org/10.1002/ajmg.a.36887.
Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–18. https://doi.org/10.1056/NEJMoa1312625.
Lo MS. Monogenic lupus. Curr Rheumatol Rep. 2016;18(12):71. https://doi.org/10.1007/s11926-016-0621-9.
Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis Res Ther. 2011;13(1):101. https://doi.org/10.1186/ar3204.
Manolio TA. Bringing genome-wide association findings into clinical use. Nat Rev Genet. 2013;14(8):549–58. https://doi.org/10.1038/nrg3523.
Hughes T, Adler A, Merrill JT, Kelly JA, Kaufman KM, Williams A, et al. Analysis of autosomal genes reveals gene-sex interactions and higher total genetic risk in men with systemic lupus erythematosus. Ann Rheum Dis. 2012;71(5):694–9. https://doi.org/10.1136/annrheumdis-2011-200385.
Webb R, Kelly JA, Somers EC, Hughes T, Kaufman KM, Sanchez E, et al. Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann Rheum Dis. 2011;70(1):151–6. https://doi.org/10.1136/ard.2010.141697.
Ceccarelli F, Perricone C, Borgiani P, Ciccacci C, Rufini S, Cipriano E, et al. Genetic factors in systemic lupus erythematosus: contribution to disease phenotype. J Immunol Res. 2015;2015:745647. https://doi.org/10.1155/2015/745647.
Kim-Howard X, Maiti AK, Anaya JM, Bruner GR, Brown E, Merrill JT, et al. ITGAM coding variant (rs1143679) influences the risk of renal disease, discoid rash and immunological manifestations in patients with systemic lupus erythematosus with European ancestry. Ann Rheum Dis. 2010;69(7):1329–32. https://doi.org/10.1136/ard.2009.120543.
Chung SA, Taylor KE, Graham RR, Nititham J, Lee AT, Ortmann WA, et al. Differential genetic associations for systemic lupus erythematosus based on anti-dsDNA autoantibody production. PLoS Genet. 2011;7(3):e1001323. https://doi.org/10.1371/journal.pgen.1001323.
Ho RC, Ong H, Thiaghu C, Lu Y, Ho CS, Zhang MW. Genetic variants that are associated with neuropsychiatric systemic lupus erythematosus. J Rheumatol. 2016;43(3):541–51. https://doi.org/10.3899/jrheum.150884.
Taylor KE, Chung SA, Graham RR, Ortmann WA, Lee AT, Langefeld CD, et al. Risk alleles for systemic lupus erythematosus in a large case-control collection and associations with clinical subphenotypes. PLoS Genet. 2011;7(2):e1001311. https://doi.org/10.1371/journal.pgen.1001311.
Chung SA, Brown EE, Williams AH, Ramos PS, Berthier CC, Bhangale T, et al. Lupus nephritis susceptibility loci in women with systemic lupus erythematosus. J Am Soc Nephrol. 2014;25(12):2859–70. https://doi.org/10.1681/ASN.2013050446.
Freedman BI, Langefeld CD, Andringa KK, Croker JA, Williams AH, Garner NE, et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. 2014;66(2):390–6. https://doi.org/10.1002/art. 38220.
Lee JC, Biasci D, Roberts R, Gearry RB, Mansfield JC, Ahmad T, et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat Genet. 2017;49(2):262–8. https://doi.org/10.1038/ng.3755.
• Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376–81. https://doi.org/10.1038/nature12873. Implement genetic findings for drug development in rheumatoid arthritis
• Finan C, Gaulton A, Kruger FA, Lumbers RT, Shah T, Engmann J, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med. 2017;9(383) https://doi.org/10.1126/scitranslmed.aag1166. Development of druggable genome to inform the design of new genotyping arrays that will enable association studies of druggable genes for drug target selection in human disease
Young KA, Munroe ME, Guthridge JM, Kamen DL, Niewold TB, Gilkeson GS, et al. Combined role of vitamin D status and CYP24A1 in the transition to systemic lupus erythematosus. Ann Rheum Dis. 2017;76(1):153–8. https://doi.org/10.1136/annrheumdis-2016-209157.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Systemic Lupus Erythematosus
Rights and permissions
About this article

Cite this article
Deng, Y., Tsao, B.P. Updates in Lupus Genetics. Curr Rheumatol Rep 19, 68 (2017). https://doi.org/10.1007/s11926-017-0695-z
Published:
DOI: https://doi.org/10.1007/s11926-017-0695-z