
Abstract
Synucleinopathies are a group of neurodegenerative diseases that share a common pathological lesion of intracellular protein inclusions largely composed by aggregates of alpha-synuclein protein. Accumulating evidence, including genome wide association studies, has implicated alpha-synuclein (SNCA) gene in the etiology of synucleinopathies. However, the precise variants within SNCA gene that contribute to the sporadic forms of Parkinson’s disease (PD), dementia with Lewy bodies (DLB), multiple system atrophy (MSA), and other synucleinopathies and their molecular mechanisms of action remain elusive. It has been suggested that SNCA expression levels are critical for the development of these diseases. Here, we review several model systems that have been developed to advance the understanding of the role of SNCA expression levels in the etiology of synucleinopathies. We also describe different molecular mechanisms that regulate SNCA gene expression and discuss possible strategies for SNCA down-regulation as means for therapeutic approaches. Finally, we highlight some examples that underscore the relationships between the genetic association findings and the regulatory mechanisms of SNCA expression, which suggest that genetic variability in SNCA locus is directly responsible, at least in part, to the changes in gene expression and explain the reported associations of SNCA with synucleinopathies. Future studies utilizing induced pluripotent stem cells (iPSCs)—derived neuronal lines and genome editing by CRISPR/Cas9, will allow us to validate, characterize, and manipulate the effects of particular cis-genetic variants on SNCA expression. Moreover, this model system will enable us to compare different neuronal and glial lineages involved in synucleinopathies representing an attractive strategy to elucidate—common and specific—SNCA-genetic variants, regulatory mechanisms, and vulnerable expression levels underlying synucleinopathy spectrum disorders. This forthcoming knowledge will support the development of precision medicine for synucleinopathies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alpha synuclein (α-syn) protein was originally identified as a precursor protein for the non-β-amyloid component (NAC) of Alzheimer’s disease plaques [1]. The α-syn, encoded by the SNCA gene, is a small (14 kD) presynaptic nerve terminal protein, abundant in the brain, and its function is not known. Parkinson’s disease (PD) and related disorders known as synucleinopathies share a common pathological lesion composed of protein inclusions in the cytoplasm of selected populations of neurons and glia (i.e., oligodendrocytes), known as Lewy bodies (LBs) and Lewy neurites, and glial cytoplasmic inclusions (GCIs), respectively [2–6]. Aggregates of the insoluble α-syn protein are the major component of LBs [7], Lewy neurites, and GCIs. In addition to PD, these groups of disorders include dementia with Lewy bodies (DLB), Alzheimer disease (AD) with Lewy bodies (LBV/AD), neurodegeneration with brain iron accumulation (NBIA) type I, pure autonomic failure (PAF), and multiple system atrophy (MSA). SNCA was the first gene implicated in familial Parkinson’s disease (fPD) [8]. Over the last decade, genome wide association studies (GWASs) and candidate gene-based approaches [9] have implicated SNCA as a highly significant genetic risk factor for synucleinopathies including sporadic PD [10–23], DLB [24], MSA [25, 26], and LBV/AD [27, 28]. However, while coding missense mutations and multiplication of SNCA locus led to the fPD [8, 29–31], the precise variants within SNCA gene that contribute to the sporadic forms of PD, DLB, MSA, and other synucleinopathies and their molecular mechanisms of action remain elusive.
The molecular mechanisms through which the SNCA gene elicits synucleinopathies and the underlying genetic factors have been studied most extensively in relation to the etiology of sporadic PD. To date, accumulating evidence has been reported in both in vitro systems and in vivo models, suggesting that the α-syn expression levels are critical for the development of the disease. In this review, we will describe recent advances in understanding the role of SNCA gene and its expression levels in sporadic PD (spPD) and other synucleinopathies. We will also discuss several regulatory mechanisms of SNCA gene expression and possible means for manipulating SNCA overexpression for therapeutic approaches.
Alpha-synuclein overexpression
The role of α-syn expression levels in disease pathogenesis has been studied in a variety of biological systems. Here, we review major approaches and current advancements.
Cellular model systems
Cell-based models, including established immortalized cell lines such as HEK293 cells [32] and SH-SY5Y neuroblastoma cells [33, 34], and primary neuronal cultures [35–37], have been widely used to study the effect of α-syn expression levels on cellular phenotypes. Although overexpression of α-syn in those mammalian cell culture systems did not show evidence for aggregation, it was shown that expression levels of α-syn are crucial for the neurodegenerative process, and it was suggested that there is a threshold level above which the detrimental function of α-syn emerge [38]. Xu et al. showed that α-syn overexpression in dopaminergic neurons leads to apoptosis, while an opposite neuroprotective effect was observed in non-dopaminergic cortical neurons. They suggested that the vulnerability of the dopaminergic cell population was associated to dopamine (DA) production [39]. It was also shown that α-syn overexpression induces oxidative stress in SH-SY5Y cells through unclear mechanisms [40]. The effect of α-syn overexpression on oligodendrocytes was evaluated in rodent oligodendroglial cell line expressing wild-type (WT) human α-syn and revealed that α-syn delays oligodendrocyte progenitor cell (OPC) maturation by severely down-regulating myelin-gene regulatory factor and myelin basic protein [41].
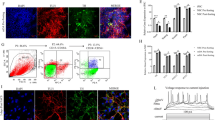
Modeling human neurodegenerative diseases by using induced pluripotent stem cells (iPSCs) leverages the use of cell culture systems in neurodegenerative research. The iPSC approach uses patient-derived cells and differentiates these cells into disease-relevant neuronal populations. In particular, the differentiation into dopaminergic and cholinergic neurons could address specific gaps in the mechanisms of neurodegeneration of PD and DLB, respectively, while, iPSC-derived oligodendrocytes represent a powerful tool for modeling MSA. The vulnerability of the dopaminergic neurons to overexpression of SNCA has been confirmed by differentiating iPSCs with SNCA triplication into dopaminergic neurons [42, 43]. The iPSC-derived dopaminergic neurons carrying the SNCA triplication showed an approximately twofold increase in α-syn levels compared to the control cells. α-syn accumulation coincided with elevated oxidative stress markers and conferred increased vulnerability to oxidative stress-induced cell death [44]. These studies introduced the iPSC-derived dopaminergic neurons as a model to investigate the effects of SNCA overexpression on PD-related phenotypes. Another study that evaluated the consequences of SNCA triplication using iPSCs, reported that iPSC-derived neural precursor cells (NPCs) displayed overall normal cellular and mitochondrial morphology but showed substantial changes in growth, viability, cellular energy metabolism, and stress resistance especially when challenged by starvation or toxicant challenge. Importantly, knockdown of SNCA overexpression resulted in reversal of the observed phenotypic changes [45]. Deriving iPSCs from both genetic and idiopathic patients could advance our understanding whether a given mutation or a particular genetic background confers selective susceptibility in a specific neuronal-type population. It has been reported recently that SNCA triplication interferes with the differentiation of iPSCs into dopaminergic and GABAergic neurons. This study showed that increased level of α-syn affects messenger RNA (mRNA) expression of genes implicated in neuronal differentiation leading to delayed maturation [46]. In unpublished work, we have developed a system to evaluate the effect of up-regulation of SNCA mRNA on cholinergic neuronal differentiation and maturation (Fig. 1). SNCA-mRNA and protein expressions in both the SNCA triplication and the control iPSC lines increased along the maturation process (Fig. 1a), and although the SNCA triplication line exhibited twofold overexpression of SNCA compared to the control cell line (Fig. 1a–c), there was no effect on the maturation process and the neurite outgrowth (Fig. 1d). A deep characterization of this cell line is underway, and the implication to DLB warrants further investigations. A new study pioneered the generation of oligodendrocytes from MSA and familial PD iPSC lines. Expression of SNCA transcript and protein was detected in those oligodendrocyte lineage cells, offering cellular models for studying the functional implication of α-syn during oligodendrocyte development and in MSA [47]. Collectively, iPSC models provide access to cell types of interest that were previously unobtainable in sufficient quantity or quality and presents exciting promises for the elucidation of synucleionopathies. However, cellular model systems lack the microenvironment (interaction with other cell type and microvasculature), occurs in the intact brain tissue, and lack the whole organism in vivo factors necessary to trigger the progression of the pathology and to evaluate phenotypic outcomes in the organism level. Furthermore, the maturation period of the differentiated neurons should be considered when establishing an iPSC model system to study neurodegenerative diseases such as synucleinopathies. For example, Crompton et al. reported that the iPSC-derived cholinergic neurons were maturated for almost 50 days when they observed neuronal specific markers that demonstrated that the cells exhibited characteristics of cholinergic neurons present in the human brains [48]. It is also important to note that iPSC-derived neurons have to undergo a prolonged maturation in culture [49], or other “aging” protocols [50, 51], in order to be comparable to the mature adult brain.

Generation of cholinergic neurons from SNCA triplication and control iPSC lines. Induced pluripotent stem cells (iPSCs) derived from a Parkinson’s disease patient with the triplication of the human SNCA genomic locus (SNCA-Tri) and from a healthy individual (control) were differentiated to basal forebrain cholinergic neurons (BFCNs). Following forebrain patterning, neural precursor cells (NPCs) were passaged at day 16+. BFCNs were next maturated for 5–7 weeks. a SNCA-mRNA expression levels at different stages of the neuronal differentiation. Levels of SNCA-mRNA were measured by real-time RT-PCR and calculated relatively to the geometric mean of GAPDH-mRNA and PPIA-mRNA reference control using the 2−ΔCT method. The blue bars represent SNCA-mRNA fold expression levels measured in the control cell lines, and the red bars represent SNCA-mRNA fold expression levels measured in the SNCA-Tri lines. Immunolabeling for α-syn protein (Alexa-488) was performed in matured cholinergic neurons (BFCNs, day 35) from control (b) and SNCA-Tri (c). Immunocytochemistry was analyzed with by confocal microscopy end fragment. d Phase-contrast micrograph of representative mature cholinergic neurons (day 50) differentiated from SNCA-Tri iPSC. The arrows indicate the neuritis outgrowth
Animal model systems
Developing animal model systems represent an in vivo approach to understand disease mechanisms. Both overexpression and down-regulation of α-syn have been modeled in several animal systems [52]. The limitation in the use of these animal models is mainly related to the difficulties in recapitulating the pathological progression and mimicking disease phenotypes. Nevertheless, animal models represent an important tool in understanding the pathogenesis mechanisms and processes leading to the disorder in a whole organism level and support the identification of novel therapeutic targets and potential therapies [53].
Caenorhabditis elegans does not contain a homolog to the human α-syn protein, providing an opportunity to explore the role of human α-syn in relation to dopaminergic neuronal vulnerability. Transgenic C. elegans have been developed, overexpressing WT and mutated α-syn in specific subsets of neurons [54]. Overexpression of WT human α-syn led to moderate dopaminergic neuronal loss.
Transgenic Drosophila melanogaster overexpressing WT or mutant human α-syn recapitulated PD-related pathological features including dopaminergic neuron loss, filamentous intraneuronal α-syn inclusions, and locomotor dysfunction [55]. These phenotypes were reverted by L-DOPA or DA agonists, suggesting the usefulness of these models for genetic screens.
Transgenic rodent models have been the most widely used animal model to study the role of SNCA gene in PD and related diseases. Several rat model systems overexpressing the human α-syn have been established and showed phenotypes closely related to those in human PD [56, 57]. Many mouse lines overexpressing human-SNCA (hSNCA) under various promoters have been created. For example, transgenic mice expressing WT hSNCA140 under the control of platelet-derived growth factor-β promoter were characterized by a reduction in tyrosine hydroxylase and DA in the striatum and progressive accumulation of α-syn and ubiquitin-immunoreactive inclusions. This model suggests that α-syn accumulation may play a causal role in PD and related conditions [58]. Mice overexpressing SNCA under the Thy1 promoter are characterized by a region-specific increase of most hSNCA transcripts (SNCA140, SNCA126, SNCA112, and SNCA98) when compared to WT mice, consistent with the region-specific elevation in hSNCA transcript level in PD patients [59]. Using the same model system, Subramaniam et al. discovered early and regionally selective alterations in mitochondrial function and oxidative stress [60]. Altered mitochondrial functions in the striatum and substantia nigra are likely due to endogenous factors that render neurons in these regions less able to cope with α-syn-induced mitochondrial dysfunction. This data is in agreement with previous findings in the postmortem substantia nigra of patients with idiopathic PD [61] but reveals, for the first time, that α-syn alters mitochondrial function to a greater extent in the regions containing nigrostriatal dopaminergic neurons than in other brain regions, specifically the cerebral cortex. Interestingly, transgenic mice overexpressing SNCA under BAC promoter show deficits in DA neurotransmission and motor impairments in the absence of α-syn aggregates, suggesting that PD-related phenotypes may be associated with much earlier deficits in DA neurotransmission [62], and possibly, the damage due to α-syn overexpression occurs earlier than the apparent accumulation of aggregates. The role of α-syn in MSA has been evaluated as well by the creation of transgenic animal models. Mice models overexpressing human WT α-syn in oligodendrocytes under the control of the 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP) promoter [63] or the proteolipid protein (PLP) promoter [64] recapitulated features of MSA such as GCI-like α-syn aggregates in oligodendrocytes, oligodendroglial and neuronal loss, and slowly progressive motor impairment.
In primates, transgenic monkeys overexpressing WT or mutant α-syn showed chronic and progressive features of human PD such as motor impairments, α-syn inclusions, and dystrophic neurites [65].
Human studies
Copy number variations [31, 66–69] in the SNCA gene have been identified in only a few families with an early-onset, autosomal dominant form of PD. Genomic triplication of the region containing SNCA results in four fully functional copies of SNCA and twofold overexpression of SNCA mRNA and protein. This leads to high penetrance of an early-onset PD phenotype with cognitive impairment and autonomic dysfunction [31, 70, 71]. Duplications of the wild-type SNCA gene result in a 1.5-fold elevation of SNCA expression and, compared with the triplication, a slightly later onset of heritable PD that is characterized by a lower penetrance and a “milder” phenotype with slower progression [66–69], demonstrating the dose-dependent effect of SNCA on disease etiology.
The role of SNCA levels was also demonstrated in the idiopathic form of sporadic PD (spPD). Elevated levels of SNCA-mRNA have been reported in midbrain tissues [72] and in dopaminergic neurons of the substantia nigra [73] from spPD postmortem brains compared to controls, suggestive of a general role for SNCA overexpression in PD. Moreover, the role of SNCA overexpression was also demonstrated in other synucleinopathies. Analysis of SNCA mRNA expression in human temporal cortex in PD and DLB demonstrated a correlation between the number of α-syn-immunoreactive LBs and the abundance of SNCA-mRNA [74]. It was also shown that oligodendrocytes isolated from MSA brains expressed elevated levels of SNCA-mRNA compared to control [75]. Recent gene expression analysis demonstrated that SNCA-mRNA levels were significantly higher in temporal cortex from LBV/AD brains compared with AD controls [27, 76]. While the studies discussed above are strongly agreeable regarding the pathogenic effect of overexpression of SNCA, other studies presented contradicting findings. For example, Neystat et al. showed a 15 to 20 % reduction of SNCA-mRNA expression in substantia nigra from PD cases relative to normal, but the same group reported no alteration in expression of SNCA-mRNA in homogenates of frontal cortex [77]. Another group reported an approximately 50 % reduction in SNCA-mRNA expression at the cellular level in substantia nigra neurons and frontal cortex neurons in PD [78]. Noteworthy, these results reflect the SNCA-mRNA of the “surviving” neurons in the disease-affected brains; therefore, we need to interpret the outcomes from gene expression analyses of neuropathological degenerative brain tissues with caution. Differences in SNCA level between PD and normal controls were evaluated in human tissues/cells other than brains as well. A mRNA expression study of peripheral lymphocytes showed no significant alteration of SNCA-mRNA expression in between spPD and controls [79]. The quantification of α-syn protein levels in blood also provided conflicting data. While some studies showed increased levels of α-syn in the plasma of PD patients compared to the controls [80], others reported decreased [81] or no significant differences [82] in plasma. These conflicting results might be attributable to the different methodologies and experimental designs of the studies and also imply a possible tissue-/cell type-specific effect. Furthermore, elevation in SNCA-mRNA levels might contribute to disease pathogenesis in many but, perhaps, not all cases of spPD.
In normal primate brains, expression of α-syn increases with age in parallel with decreased tyrosine hydroxylase expression [83]. Accordingly, some investigators have hypothesized that PD and related diseases represent accelerated variants of the normal aging process, where inappropriate degradation or overexpression of α-syn plays a central role [84, 85]. This observation provided further support to the pathogenic effect of SNCA overexpression.
Intervention approaches for SNCA down-regulation and the impact
Specific targeting of α-syn expression levels represents an attractive neuroprotective strategy, and manipulations of SNCA levels have had beneficial effects. RNA interference (RNAi) approach has been tested in vitro in neuron-like cell cultures and in vivo in rodent and other animal models [86–89]. Different effects of RNAi strategies have been reported in different cell lines. Down-regulation of SNCA in MN9D cells decreases cell viability [89]. Two adeno-associated virus (AAV) gene-silencing vectors have also been tested for efficiency and specificity of silencing and toxicity in 293T, PC12, and SH-SY5Y cells. One vector was embedded in a microRNA backbone while the second was not [90]. Both vectors silenced hSNCA to the same extent and with high specificity. However, the mir30-embedded vector was significantly less toxic [90]. Knockdown of SNCA in a triplication NPCs by short hairpin RNA (shRNA) resulted in reversal of the observed phenotypic abnormalities in growth, viability, cellular energy metabolism, and stress resistance [45]. Knocking down SNCA levels was also evaluated in animal models. siRNAs against SNCA injected directly into the monkey substantia nigra achieved over 50 % down-regulation of expression in nigrostriatal dopaminergic neurons [91]. A new siRNA approach, “expression-control RNAi” (ExCont-RNAi) treatment of PD flies, demonstrated a significant improvement in their motor function [92]. This study suggested that α-syn overexpression is associated with the degree of motor dysfunction and that it is possible to determine a threshold between benign and malignant levels of α-syn. shRNA strategies have been used in rat models with both lentiviral [87] and AAV deliveries [88]. Lentiviral delivery was able to silence specifically ectopic α-syn expression, and no toxicity was reported as a consequence of these treatments. The use of AAV vectors caused a significant loss of nigrostriatal dopaminergic neurons. However, there was also neurotoxicity associated with robust reduction of SNCA levels mediated by siRNA in those rat models [88, 93].
In conclusion, the role of SNCA overexpression in PD pathogenesis on the one hand, and the need to maintain normal physiological levels of α-syn protein on the other, emphasizes the so-far unmet need to determine the threshold between physiological and pathological α-syn levels and underscores the importance of understanding the mechanisms and factors that participate in the expression regulation of the SNCA gene.
Regulatory mechanisms of SNCA gene expression
Transcriptional and post-transcriptional mechanisms regulate SNCA gene expression and could have an important role in the development of synucleinopathies. Here, we describe the current knowledge of molecular mechanisms that control SNCA expression levels.
Transcription
Several groups have studied the transcription regulation of SNCA and have identified few putative transcription factors (TFs) that mediate SNCA expression. We identified the transcriptional activator, poly(ADP-ribose) transferase/polymerase-1 (PARP-1), that binds to Rep1 and regulates SNCA expression via this interaction [94]. Transcription factors of the GATA family [95] and ZSCAN21 [96] bind to elements within intron 1 and the promoter [97] region of SNCA and have been proposed to play a role as inducers of transcription. Clough et al. suggested that a signaling pathway involving ERK/PI3-mediated ZSCAN induced SNCA transcriptional activation [96, 98, 99]. Sterling et al. identified cis-regulating, evolutionarily conserved genomic elements in the SNCA locus that modulated the expression of a reporter gene and found five factors (PITX3, OTX2, NR3C1, AR, and TBP) that interacted with the SNCA promoter [100]. Additional regulatory areas may be located outside the promoter regions, such as intronic or intergenic regions [100]. The regulatory role of intronic regions has been confirmed by combining in silico and wet bench approaches that led to the identification of an intronic cytosine-thymine (CT)-rich region in SNCA gene that influences SNCA transcript levels [76].
microRNAs
Modulation of SNCA-mRNA levels by endogenous microRNAs (miRNAs) was proposed a post-transcriptional mechanism of SNCA regulation. Two miRNAs—miR-7 and miR-153—that are abundantly expressed in the brain have been implicated in the regulation of SNCA transcript levels. In rodent primary neurons, both miR-7 and miR-153 down-regulated SNCA levels and showed an additive effect [101]. In PD human brains, miR-34b and miR-34c are down-regulated [102, 103]. The effect of these miRNAs has been investigated in SH-SY5Y cells. Both miR-34b and miR-34c decrease SNCA-mRNA levels and α-syn protein by targeting the 3′ untranslated region (3′UTR) of SNCA mRNA [104].
Splicing
Alternative splicing is another post-transcriptional mechanism that regulates expression of SNCA transcripts. At least six different SNCA transcript variants have been described for SNCA gene, SNCA140, SNCA126, SNCA115, SNCA112, SNCA98, and SNCA67 [59, 105], of which SNCA112, SNCA126, and SNCA98 arise from alternative splicing. No one has yet discovered the biological and pathological significance of the different SNCA isoforms. However, specific isoforms have been associated with intracellular aggregation [106] and are differently expressed in human synucleinopathies [107]. A deletion of either exon 3 or 5 predicts functional consequences; while exon 3 deletion (SNCA126) leads to the interruption of the N-terminal protein-membrane interaction domain which may lead to less aggregation, exon 5 deletion (SNCA112) may result in enhanced aggregation due to a significant shortening of the unstructured C-terminus [108, 109]. In frontal cortex of DLB, SNCA112 is increased markedly compared to the controls [110], while SNCA126 levels are decreased in the prefrontal cortex of DLB patients [111]. In contrast, SNCA126 expression showed increased in the frontal cortex of PD brains and no significant differences in MSA [107]. SNCA98 is a brain-specific splice variant that lacks both exons 3 and 5 and exhibits different expression levels in various areas of fetal and adult brains. Overexpression of SNCA98 has been reported in DLB, PD [112], and MSA [107] frontal cortices compared with controls.
Post-translation
Post-translational degradation also regulates α-syn protein levels; however, the specific degradation pathway is still controversial. It was suggested that α-syn clearance is related to α-syn levels, its assembly state, and to the homeostatic environment in which degradation occurs. The ubiquitin-proteasome system (UPS) and the autophagy lysosome pathway (ALP) influence α-syn turnover as well [113]. Interestingly, aging affects these pathways, leading to α-syn accumulation [114]. Increased α-syn levels promote the generation of aberrant species that impair UPS and ALP determining a bidirectional positive feedback loop, leading to neuronal death [115].
In this view, tight regulation of the expression levels of SNCA, mainly through the various transcriptional and post-transcriptional pathways, presents a promising avenue to fine-tune α-syn levels to the degree required for successful therapeutic intervention. Thus, continued investigations of the molecular mechanisms and trans-acting factors that regulate SNCA expression in a cell type-specific manner—neuronal (dopaminergic and cortical) and oligodendrocyte—and identification of their corresponding binding sequence elements will have a high impact on the development of therapeutic targets.
Genetic and epigenetic regulation of SNCA expression
Differential regulation of SNCA expression can be attributed, in part, to genetic variability across the SNCA genomic region and intergenic sequences. We highlight several examples below:
-
A polymorphic dinucleotide complex repeat site located ∼10 kb upstream of the SNCA transcription start site [116, 117], named Rep1, has been described [118]. The length of Rep1 appears to be associated with increased risk of PD [119]. The overwhelming majority of the reported association studies, including a large meta-analysis, have shown that the extended alleles of SNCA-Rep1 confer increased risk to develop late-onset, “idiopathic” PD, while the shorter allele is protective [20, 120–123]. We investigated the effect of the Rep1 polymorphism on SNCA expression and discovered that Rep1 regulates SNCA transcription in human brain tissues. These results have been confirmed using luciferase reporter assay and in a humanized mouse model. The PD risk Rep1 allele led to increased SNCA-mRNA levels, providing further support to the pathogenic effect of SNCA overexpression [118, 124, 125]. In support of this, we identified a factor, PARP-1, that binds specifically to Rep1 and modulates SNCA transcription [94], suggestion that the association of Rep1 alleles with sporadic PD may be mediated, in part, by the effect of PARP-1 on SNCA expression.
-
Recently, four distinct haplotypes within a highly polymorphic low-complexity CT-rich region in intron 4 of SNCA gene were identified. A specific haplotype conferred risk to develop LBV/AD. It was further demonstrated that the CT-rich site acts as an enhancer element, where the risk haplotype was significantly associated with elevated levels of SNCA mRNA [76].
-
The SNCA126 splicing variant may have a protective role. A poly-T variant in intron 2 of SNCA gene comprises three alleles (5T, 7T, and 12T), and the length of the poly-T stretch is directly associated with SNCA126 expression levels in the normal brain, influencing the splicing efficiency of SNCA exon 3. Whereas the shortest 5T allele was associated with lower expression of SNCA126-mRNA, the longest 12T led to the highest SNCA126-mRNA levels [126]. The same study also reported that the 12T allele-carrying genotypes accumulated with increasing age in the normal population, while the frequency of 5T allele-carrying genotypes decreased in successive age groups until reaching zero in the oldest group. Collectively, these observations imply that the longest poly-T allele has a protective effect in aging, presumably via its association with higher SNCA126-mRNA levels.
-
Single-nucleotide polymorphisms (SNPs) tagging the SNCA 3′ showed significant effects on the relative levels of SNCA112-mRNA (exon 5 in-frame skipping) from total SNCA transcript levels in human brain tissues. The reported disease risk alleles were correlated with increased expression ratio of SNCA112-mRNA from total SNCA-mRNA [127]. Interestingly, it has been suggested that exon 5 deletion (SNCA112) results in enhanced α-syn aggregation due to a significant shortening of the unstructured C-terminus [108, 109]. This study provided evidence for functional consequences of PD-associated SNCA gene variants at the 3′ region, suggesting that genetic regulation of SNCA splicing plays an important role in the development of the disease. A follow-up analysis using bioinformatic tools found potential splicing enhancer/silencer cis-elements within the sequences surrounding the SNPs and identified SR protein-binding motifs (in particular for SRp40) that might be created or disrupted by these SNPs. Further empirical studies to determine the definite functional variant/s within SNCA 3′ and to establish their association with PD pathology are necessary [128].
-
The minor allele of SNP rs17016074 at the SNCA 3′UTR increased luciferase expression in SH-SY5Y neuroblastoma cells. SNPs in the 3′UTR including rs17016074 were associated with increased PD risk. These findings demonstrated that the 3′UTR of human SNCA, as a whole, and rs17016074, in particular, are loci of potential importance for disease development possibly via post-transcriptional effect on SNCA expression levels [128].
-
SNP rs10024743 in SNCA 3′UTR lies within a target site for miR-34b and was found to lower the miR-34b-mediated repression of the α-syn protein. This study suggested that down-regulation of miR-34b and miR-34c in the brain, as well as a SNP in the 3′UTR of SNCA gene, can increase α-syn expression, possibly contributing to PD pathogenesis [104].
GWAS, candidate gene-based studies, and meta-analyses [9, 14, 20, 24–26, 121, 129–134] showed that polymorphisms of SNCA are associated with spPD, DLB, MSA, and other synucleinopathies. The examples above demonstrated that non-coding genetic variants in SNCA genomic region contribute to the development of synucleinopathies possibly via cis-regulation of the gene expression and suggested that changes in SNCA expression profile is the molecular mechanism that mediates the reported genetic associations.
Epigenetic regulation of SNCA expression, in particular DNA methylation, was suggested to play a key role [135]. However, there are conflicting reports regarding the link between DNA methylation and PD pathogenesis. It was shown that methylation of intron 1 decreased SNCA transcription [136, 137]. Interestingly, in PD and DLB human brains, reduced DNA methylation has been reported, leading to increased α-syn expression [136, 137]. Conversely, no differences have been reported by comparing DNA methylation levels of CpG islands in SNCA intron 1 between normal and PD leukocytes [138].
Conclusion remarks
We reported experimental evidence, from different disciplines, of the role of α-syn overexpression in the pathogenesis of synucleinopathies in general and PD in detail. Herein, we propose a model, whereas even subtle changes in the α-syn expression are able to trigger the onset of synucleinopathies. The changes in the α-syn expression can be attributed to different causes as outlined in Fig. 2 and are potentially cell type-specific. Built on evidence described in this review, the upper panel of Fig. 2 summarizes possible genetic mechanisms—cis-genetic factors and trans-acting modulators—leading to alter regulation of SNCA expression. This review focused on genetic regulation of gene expression and epigenomic to some extent; however, other factors including, environmental, aging, and gender may also contribute to changes in α-syn levels. The schematic model presents several plausible mechanisms of regulation of gene expression and cell type-specific threshold of vulnerability to SNCA overexpression that may underlie the commonality and differences among synucleinopathies (Fig. 2). Interestingly, the genetic associations of SNCA gene with PD and MSA are distinct from the reported association of SNCA with DLB. Consequently, the relation of the SNCA gene with the different diseases in the broader spectrum of synucleinopathies might represent a case of allelic heterogeneity and pleiotropy. This implies that common and distinct regulatory mechanism of SNCA gene expression might be involved in the etiology of synucleinopathies.

A schematic model of the contribution of SNCA expression to synucleinopathies. Changes in the SNCA expression can be attributed to different factors. The upper panel shows the cis-genetic variants across the SNCA genomic locus and their interactions with trans-acting factors, including transcription and splicing machineries and microRNAs. The middle panel presents plausible factors affecting the endpoint cellular level of α-syn protein. These factors include, but not limited to, genetic, epigenetic, environment, gender, and age and may also interact with one another and cross-influence each other. The thresholds among physiological and pathological levels of SNCA, designated in gray vertical lines, need to be determined. The thresholds of SNCA expression levels leading to the different synucleinopathy diseases could be cell type and disease specific. Dopaminergic neurons, cortical neurons (mainly cholinergic), and oligodendrocytes, indicated in the lower panel, may exhibit different vulnerability to α-syn overexpression. In addition, interactions with other causal genes may determine the particular disease path. Collectively, the model suggests that common and distinct regulatory mechanisms and vulnerability thresholds of SNCA gene expression underlie the etiology of synucleinopathies
It is widely agreeable that reducing α-syn levels represents an attractive strategy to counteract the detrimental effect of the overexpression. However, there are major gaps in the current knowledge, including the precise causal genetic variants and their mechanisms of action underlying the different synucleinopathies, the functional role of the different SNCA transcript variants in the pathogenesis of synucleionopathies, and the expression threshold above which α-syn acquires a toxic effect that lead to the initiation of the disease processes. Emerging technologies and novel model systems, including iPSC-derived neurons and genome editing by CRISPR/Cas9, will support advancements to fill in those gaps in knowledge that in return will result in the development of precision medicine, including biomarkers for preclinical diagnosis and effective treatment approaches for synucleinopathies in general and to each disease in particular.
References
Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 90:11282–11286
Galvin JE, Lee VM, Trojanowski JQ (2001) Synucleinopathies: clinical and pathological implications. Arch Neurol 58:186–190
Marti MJ, Tolosa E, Campdelacreu J (2003) Clinical overview of the synucleinopathies. Mov Disord 18(Suppl 6):S21–S27. doi:10.1002/mds.10559
Spillantini MG (1999) Parkinson’s disease, dementia with Lewy bodies and multiple system atrophy are alpha-synucleinopathies. Parkinsonism Relat Disord 5:157–162
Jellinger KA (2003) Neuropathological spectrum of synucleinopathies. Mov Disord 18(Suppl 6):S2–12. doi:10.1002/mds.10557
Goedert M, Jakes R, Anthony Crowther R, Grazia Spillantini M (2001) Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy as alpha-synucleinopathies. Methods Mol Med 62:33–59. doi:10.1385/1-59259-142-6:33
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Han W, Liu Y, Mi Y, Zhao J, Liu D, Tian Q (2015) Alpha-synuclein (SNCA) polymorphisms and susceptibility to Parkinson’s disease: a meta-analysis. Am J Med Genet B Neuropsychiatr Genet 168B:123–134. doi:10.1002/ajmg.b.32288
Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH, Psg P, GenePd Investigators C, Molecular Genetic L (2009) Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 124:593–605. doi:10.1007/s00439-008-0582-9
Myhre R, Toft M, Kachergus J, Hulihan MM, Aasly JO, Klungland H, Farrer MJ (2008) Multiple alpha-synuclein gene polymorphisms are associated with Parkinson’s disease in a Norwegian population. Acta Neurol Scand 118:320–327
Ross OA, Gosal D, Stone JT, Lincoln SJ, Heckman MG, Irvine GB, Johnston JA, Gibson JM, Farrer MJ, Lynch T (2007) Familial genes in sporadic disease: common variants of alpha-synuclein gene associate with Parkinson’s disease. Mech Ageing Dev 128:378–382
Pals P, Lincoln S, Manning J, Heckman M, Skipper L, Hulihan M, Van den Broeck M, De Pooter T, Cras P, Crook J, Van Broeckhoven C, Farrer MJ (2004) Alpha-synuclein promoter confers susceptibility to Parkinson’s disease. Ann Neurol 56:591–595
Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T, Berg D, Wullner U, Meitinger T, Gasser T (2005) Multiple regions of alpha-synuclein are associated with Parkinson’s disease. Ann Neurol 57:535–541
Mizuta I, Satake W, Nakabayashi Y, Ito C, Suzuki S, Momose Y, Nagai Y, Oka A, Inoko H, Fukae J, Saito Y, Sawabe M, Murayama S, Yamamoto M, Hattori N, Murata M, Toda T (2006) Multiple candidate gene analysis identifies alpha-synuclein as a susceptibility gene for sporadic Parkinson’s disease. Hum Mol Genet 15:1151–1158
Winkler S, Hagenah J, Lincoln S, Heckman M, Haugarvoll K, Lohmann-Hedrich K, Kostic V, Farrer M, Klein C (2007) {alpha}-synuclein and Parkinson disease susceptibility. Neurology 69:1745–50
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41:1303–1307
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41:1308–1312
Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Zuchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER (2010) Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. doi:10.1111/j.1469-1809.2009.00560.x
Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven C (2006) Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. Jama 296:661–670
CC S, Plagnol V, Strange A, Gardner M, Paisan-Ruiz C, Band G, RA B, Bellenguez C, Bhatia K, Blackburn H, JM B, Bramon E, MA B, Burn D, JP C, PF C, CE C, Corvin A, Craddock N, Deloukas P, Edkins S, Evans J, Freeman C, Gray E, Hardy J, Hudson G, Hunt S, Jankowski J, Langford C, AJ L, HS M, CG M, MI MC, KE M, CN P, JP P, Peltonen L, Pirinen M, Plomin R, Potter S, Rautanen A, SJ S, Z S, RC T, AC V, NW W, HR M, Donnelly P, NW W (2010) Dissection of the genetics of Parkinson’s disease identifies an additional association 5’ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet 20:345–353. doi:10.1093/hmg/ddq469
Simon-Sanchez J, van Hilten JJ, van de Warrenburg B, Post B, Berendse HW, Arepalli S, Hernandez DG, de Bie RM, Velseboer D, Scheffer H, Bloem B, van Dijk KD, Rivadeneira F, Hofman A, Uitterlinden AG, Rizzu P, Bochdanovits Z, Singleton AB, Heutink P (2011) Genome-wide association study confirms extant PD risk loci among the Dutch. Eur J Hum Genet. doi:10.1038/ejhg.2010.254
Mata IF, Shi M, Agarwal P, Chung KA, Edwards KL, Factor SA, Galasko DR, Ginghina C, Griffith A, Higgins DS, Kay DM, Kim H, Leverenz JB, Quinn JF, Roberts JW, Samii A, Snapinn KW, Tsuang DW, Yearout D, Zhang J, Payami H, Zabetian CP (2010) SNCA variant associated with Parkinson disease and plasma alpha-synuclein level. Arch Neurol 67:1350–1356. doi:10.1001/archneurol.2010.279
Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O, Escott-Price V, Hernandez DG, Nalls MA, Clark LN, Honig LS, Marder K, Van Der Flier WM, Lemstra A, Scheltens P, Rogaeva E, St George-Hyslop P, Londos E, Zetterberg H, Ortega-Cubero S, Pastor P, Ferman TJ, Graff-Radford NR, Ross OA, Barber I, Braae A, Brown K, Morgan K, Maetzler W, Berg D, Troakes C, Al-Sarraj S, Lashley T, Compta Y, Revesz T, Lees A, Cairns N, Halliday GM, Mann D, Pickering-Brown S, Dickson DW, Singleton A, Hardy J (2014) Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet 23:6139–6146. doi:10.1093/hmg/ddu334
Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, Paisan-Ruiz C, Bras J, Ding J, Chen H, Traynor BJ, Arepalli S, Zonozi RR, Revesz T, Holton J, Wood N, Lees A, Oertel W, Wullner U, Goldwurm S, Pellecchia MT, Illig T, Riess O, Fernandez HH, Rodriguez RL, Okun MS, Poewe W, Wenning GK, Hardy JA, Singleton AB, Gasser T (2009) SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 65:610–614
Al-Chalabi A, Durr A, Wood NW, Parkinson MH, Camuzat A, Hulot JS, Morrison KE, Renton A, Sussmuth SD, Landwehrmeyer BG, Ludolph A, Agid Y, Brice A, Leigh PN, Bensimon G, Group NGS (2009) Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy. PLoS one 4:e7114. doi:10.1371/journal.pone.0007114
Linnertz C, Lutz MW, Ervin JF, Allen J, Miller NR, Welsh-Bohmer KA, Roses AD, Chiba-Falek O (2014) The genetic contributions of SNCA and LRRK2 genes to Lewy body pathology in Alzheimer’s disease. Hum Mol Genet 23:4814–4821. doi:10.1093/hmg/ddu196
Lutz MW, Saul R, Linnertz C, Glenn OC, Roses AD, Chiba-Falek O (2015) A cytosine-thymine (CT)-rich haplotype in intron 4 of SNCA confers risk for Lewy body pathology in Alzheimer’s disease and affects SNCA expression. Alzheimer’s Dementia: J Alzheimer’s Assoc. doi:10.1016/j.jalz.2015.05.011
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18:106–108
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) Alpha-synuclein locus triplication causes Parkinson’s disease. Science 302:841
Tabrizi SJ, Orth M, Wilkinson JM, Taanman JW, Warner TT, Cooper JM, Schapira AH (2000) Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity. Hum Mol Genet 9:2683–2689
Banerjee K, Munshi S, Sen O, Pramanik V, Roy Mukherjee T, Chakrabarti S (2014) Dopamine cytotoxicity involves both oxidative and nonoxidative pathways in SH-SY5Y cells: potential role of alpha-synuclein overexpression and proteasomal inhibition in the etiopathogenesis of Parkinson’s disease. Parkinsons Dis 2014:878935. doi:10.1155/2014/878935
Vekrellis K, Xilouri M, Emmanouilidou E, Stefanis L (2009) Inducible over-expression of wild type alpha-synuclein in human neuronal cells leads to caspase-dependent non-apoptotic death. J Neurochem 109:1348–1362. doi:10.1111/j.1471-4159.2009.06054.x
Zhou W, Hurlbert MS, Schaack J, Prasad KN, Freed CR (2000) Overexpression of human alpha-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res 866:33–43
Taguchi K, Watanabe Y, Tsujimura A, Tatebe H, Miyata S, Tokuda T, Mizuno T, Tanaka M (2014) Differential expression of alpha-synuclein in hippocampal neurons. PLoS one 9:e89327. doi:10.1371/journal.pone.0089327
Koch JC, Bitow F, Haack J, d’Hedouville Z, Zhang JN, Tonges L, Michel U, Oliveira LM, Jovin TM, Liman J, Tatenhorst L, Bahr M, Lingor P (2015) Alpha-synuclein affects neurite morphology, autophagy, vesicle transport and axonal degeneration in CNS neurons. Cell Death Dis 6:e1811. doi:10.1038/cddis.2015.169
Seo JH, Rah JC, Choi SH, Shin JK, Min K, Kim HS, Park CH, Kim S, Kim EM, Lee SH, Lee S, Suh SW, Suh YH (2002) Alpha-synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J 16:1826–1828. doi:10.1096/fj.02-0041fje
Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA (2002) Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med 8:600–606. doi:10.1038/nm0602-600
Junn E, Mouradian MM (2002) Human alpha-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci Lett 320:146–150
May VE, Ettle B, Poehler AM, Nuber S, Ubhi K, Rockenstein E, Winner B, Wegner M, Masliah E, Winkler J (2014) Alpha-synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol Aging 35:2357–2368. doi:10.1016/j.neurobiolaging.2014.02.028
Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, Schapira AH, Gwinn K, Hardy J, Lewis PA, Kunath T (2011) Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun 2:440. doi:10.1038/ncomms1453
Byers B, Cord B, Nguyen HN, Schule B, Fenno L, Lee PC, Deisseroth K, Langston JW, Pera RR, Palmer TD (2011) SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS one 6:e26159. doi:10.1371/journal.pone.0026159
Byers B, Lee HL, Reijo Pera R (2012) Modeling Parkinson’s disease using induced pluripotent stem cells. Current Neurol Neurosci Rep 12:237–242. doi:10.1007/s11910-012-0270-y
Flierl A, Oliveira LM, Falomir-Lockhart LJ, Mak SK, Hesley J, Soldner F, Arndt-Jovin DJ, Jaenisch R, Langston JW, Jovin TM, Schule B (2014) Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLoS one 9:e112413. doi:10.1371/journal.pone.0112413
Oliveira LM, Falomir-Lockhart LJ, Botelho MG, Lin KH, Wales P, Koch JC, Gerhardt E, Taschenberger H, Outeiro TF, Lingor P, Schule B, Arndt-Jovin DJ, Jovin TM (2015) Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis 6:e1994. doi:10.1038/cddis.2015.318
Djelloul M, Holmqvist S, Boza-Serrano A, Azevedo C, Yeung MS, Goldwurm S, Frisen J, Deierborg T, Roybon L (2015) Alpha-synuclein expression in the oligodendrocyte lineage: an in vitro and in vivo study using rodent and human models. Stem Cell Reports 5:174–184. doi:10.1016/j.stemcr.2015.07.002
Crompton LA, Byrne ML, Taylor H, Kerrigan TL, Bru-Mercier G, Badger JL, Barbuti PA, Jo J, Tyler SJ, Allen SJ, Kunath T, Cho K, Caldwell MA (2013) Stepwise, non-adherent differentiation of human pluripotent stem cells to generate basal forebrain cholinergic neurons via hedgehog signaling. Stem Cell Res 11:1206–1221. doi:10.1016/j.scr.2013.08.002
Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, Jimenez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, Canals JM, Memo M, Alberch J, Lopez-Barneo J, Vila M, Cuervo AM, Tolosa E, Consiglio A, Raya A (2012) Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 4:380–395. doi:10.1002/emmm.201200215
Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Yanai H, Fraifeld VE (2013) The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res Rev 12:661–684. doi:10.1016/j.arr.2012.02.001
Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247. doi:10.1038/35041687
Le W, Sayana P, Jankovic J (2014) Animal models of Parkinson’s disease: a gateway to therapeutics? Neurotherapeutics 11:92–110. doi:10.1007/s13311-013-0234-1
Deng H, Yuan L (2014) Genetic variants and animal models in SNCA and Parkinson disease. Ageing Res Rev 15:161–176. doi:10.1016/j.arr.2014.04.002
Lakso M, Vartiainen S, Moilanen AM, Sirvio J, Thomas JH, Nass R, Blakely RD, Wong G (2003) Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem 86:165–172
Feany MB, Bender WW (2000) A Drosophila model of Parkinson’s disease. Nature 404:394–398
Yamada M, Iwatsubo T, Mizuno Y, Mochizuki H (2004) Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson’s disease. J Neurochem 91:451–461. doi:10.1111/j.1471-4159.2004.02728.x
Nuber S, Harmuth F, Kohl Z, Adame A, Trejo M, Schonig K, Zimmermann F, Bauer C, Casadei N, Giel C, Calaminus C, Pichler BJ, Jensen PH, Muller CP, Amato D, Kornhuber J, Teismann P, Yamakado H, Takahashi R, Winkler J, Masliah E, Riess O (2013) A progressive dopaminergic phenotype associated with neurotoxic conversion of alpha-synuclein in BAC-transgenic rats. Brain 136:412–432. doi:10.1093/brain/aws358
Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000) Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287:1265–1269
McLean JR, Hallett PJ, Cooper O, Stanley M, Isacson O (2012) Transcript expression levels of full-length alpha-synuclein and its three alternatively spliced variants in Parkinson’s disease brain regions and in a transgenic mouse model of alpha-synuclein overexpression. Mol Cell Neurosci 49:230–239. doi:10.1016/j.mcn.2011.11.006
Subramaniam SR, Vergnes L, Franich NR, Reue K, Chesselet MF (2014) Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol Dis 70:204–213. doi:10.1016/j.nbd.2014.06.017
Keeney PM, Xie J, Capaldi RA, Bennett JP Jr (2006) Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci 26:5256–5264. doi:10.1523/JNEUROSCI.0984-06.2006
Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, Parkkinen L, Senior SL, Anwar S, Ryan B, Deltheil T, Kosillo P, Cioroch M, Wagner K, Ansorge O, Bannerman DM, Bolam JP, Magill PJ, Cragg SJ, Wade-Martins R (2013) Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A 110:E4016–E4025. doi:10.1073/pnas.1309143110
Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, Trojanowski JQ, Lee VM (2005) Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 45:847–859. doi:10.1016/j.neuron.2005.01.032
Stefanova N, Reindl M, Neumann M, Haass C, Poewe W, Kahle PJ, Wenning GK (2005) Oxidative stress in transgenic mice with oligodendroglial alpha-synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. Am J Pathol 166:869–876
Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Bjorklund A (2003) Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson’s disease. Proc Natl Acad Sci U S A 100:2884–2889. doi:10.1073/pnas.0536383100
Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson EM, Schule B, Langston JW, Middleton FA, Ross OA, Hulihan M, Gasser T, Farrer MJ (2007) Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 68:916–922
Ross OA, Braithwaite AT, Skipper LM, Kachergus J, Hulihan MM, Middleton FA, Nishioka K, Fuchs J, Gasser T, Maraganore DM, Adler CH, Larvor L, Chartier-Harlin MC, Nilsson C, Langston JW, Gwinn K, Hattori N, Farrer MJ (2008) Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol 63:743–750
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364:1167–1169
Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364:1169–1171
Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW (2004) Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 55:174–179
Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, Singleton AB (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62:1835–1838
Chiba-Falek O, Lopez GJ, Nussbaum RL (2006) Levels of alpha-synuclein mRNA in sporadic Parkinson disease patients. Mov Disord 21:1703–1708. doi:10.1002/mds.21007
Grundemann J, Schlaudraff F, Haeckel O, Liss B (2008) Elevated alpha-synuclein mRNA levels in individual UV-laser-microdissected dopaminergic substantia nigra neurons in idiopathic Parkinson’s disease. Nucleic Acids Res 36:e38
Rockenstein E, Hansen LA, Mallory M, Trojanowski JQ, Galasko D, Masliah E (2001) Altered expression of the synuclein family mRNA in Lewy body and Alzheimer’s disease. Brain Res 914:48–56
Asi YT, Simpson JE, Heath PR, Wharton SB, Lees AJ, Revesz T, Houlden H, Holton JL (2014) Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia 62:964–970. doi:10.1002/glia.22653
Lutz MW, Saul R, Linnertz C, Glenn OC, Roses AD, Chiba-Falek O (2015) A cytosine-thymine (CT)-rich haplotype in intron 4 of SNCA confers risk for Lewy body pathology in Alzheimer’s disease and affects SNCA expression. Alzheimers Dement 11:1133–1143. doi:10.1016/j.jalz.2015.05.011
Neystat M, Lynch T, Przedborski S, Kholodilov N, Rzhetskaya M, Burke RE (1999) Alpha-synuclein expression in substantia nigra and cortex in Parkinson’s disease. Mov Disord 14:417–422
Kingsbury AE, Daniel SE, Sangha H, Eisen S, Lees AJ, Foster OJ (2004) Alteration in alpha-synuclein mRNA expression in Parkinson’s disease. Mov Disord 19:162–170
Tan EK, Chandran VR, Fook-Chong S, Shen H, Yew K, Teoh ML, Yuen Y, Zhao Y (2005) Alpha-synuclein mRNA expression in sporadic Parkinson’s disease. Mov Disord 20:620–623
Lee PH, Lee G, Park HJ, Bang OY, Joo IS, Huh K (2006) The plasma alpha-synuclein levels in patients with Parkinson’s disease and multiple system atrophy. J Neural Transm (Vienna) 113:1435–1439. doi:10.1007/s00702-005-0427-9
Li QX, Mok SS, Laughton KM, McLean CA, Cappai R, Masters CL, Culvenor JG, Horne MK (2007) Plasma alpha-synuclein is decreased in subjects with Parkinson’s disease. Exp Neurol 204:583–588. doi:10.1016/j.expneurol.2006.12.006
Shi M, Zabetian CP, Hancock AM, Ginghina C, Hong Z, Yearout D, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Leverenz JB, Zhang J (2010) Significance and confounders of peripheral DJ-1 and alpha-synuclein in Parkinson’s disease. Neurosci Lett 480:78–82. doi:10.1016/j.neulet.2010.06.009
Chu Y, Kordower JH (2007) Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol Dis 25:134–149. doi:10.1016/j.nbd.2006.08.021
Obeso JA (2010) Modeling clinical features of neurodegeneration. Nat Med 16:1372. doi:10.1038/nm1210-1372b
Collier TJ, Kanaan NM, Kordower JH (2011) Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat Rev Neurosci 12:359–366. doi:10.1038/nrn3039
Fountaine TM, Venda LL, Warrick N, Christian HC, Brundin P, Channon KM, Wade-Martins R (2008) The effect of alpha-synuclein knockdown on MPP+ toxicity in models of human neurons. Eur J Neurosci 28:2459–2473. doi:10.1111/j.1460-9568.2008.06527.x
Lewis J, Melrose H, Bumcrot D, Hope A, Zehr C, Lincoln S, Braithwaite A, He Z, Ogholikhan S, Hinkle K, Kent C, Toudjarska I, Charisse K, Braich R, Pandey RK, Heckman M, Maraganore DM, Crook J, Farrer MJ (2008) In vivo silencing of alpha-synuclein using naked siRNA. Mol Neurodegener 3:19
Gorbatyuk OS, Li S, Nash K, Gorbatyuk M, Lewin AS, Sullivan LF, Mandel RJ, Chen W, Meyers C, Manfredsson FP, Muzyczka N (2010) In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol Ther J. Am Soc Gene Ther 18:1450–1457. doi:10.1038/mt.2010.115
Liu D, Jin L, Wang H, Zhao H, Zhao C, Duan C, Lu L, Wu B, Yu S, Chan P, Li Y, Yang H (2008) Silencing alpha-synuclein gene expression enhances tyrosine hydroxylase activity in MN9D cells. Neurochem Res 33:1401–1409. doi:10.1007/s11064-008-9599-7
Han Y, Khodr CE, Sapru MK, Pedapati J, Bohn MC (2011) A microRNA embedded AAV alpha-synuclein gene silencing vector for dopaminergic neurons. Brain Res 1386:15–24. doi:10.1016/j.brainres.2011.02.041
McCormack AL, Mak SK, Henderson JM, Bumcrot D, Farrer MJ, Di Monte DA (2010) Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS one 5:e12122. doi:10.1371/journal.pone.0012122
Takahashi M, Suzuki M, Fukuoka M, Fujikake N, Watanabe S, Murata M, Wada K, Nagai Y, Hohjoh H (2015) Normalization of overexpressed alpha-synuclein causing Parkinson’s disease by a moderate gene silencing with RNA interference. Molecular Therapy Nucleic Acids 4:e241. doi:10.1038/mtna.2015.14
Khodr CE, Sapru MK, Pedapati J, Han Y, West NC, Kells AP, Bankiewicz KS, Bohn MC (2011) An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson’s disease, but displays toxicity in dopamine neurons. Brain Res 1395:94–107. doi:10.1016/j.brainres.2011.04.036
Chiba-Falek O, Kowalak JA, Smulson ME, Nussbaum RL (2005) Regulation of alpha-synuclein expression by poly (ADP ribose) polymerase-1 (PARP-1) binding to the NACP-Rep1 polymorphic site upstream of the SNCA gene. Am J Hum Genet 76:478–492
Scherzer CR, Grass JA, Liao Z, Pepivani I, Zheng B, Eklund AC, Ney PA, Ng J, McGoldrick M, Mollenhauer B, Bresnick EH, Schlossmacher MG (2008) GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proc Natl Acad Sci U S A 105:10907–10912. doi:10.1073/pnas.0802437105
Clough RL, Dermentzaki G, Stefanis L (2009) Functional dissection of the alpha-synuclein promoter: transcriptional regulation by ZSCAN21 and ZNF219. J Neurochem 110:1479–1490. doi:10.1111/j.1471-4159.2009.06250.x
Brenner S, Wersinger C, Gasser T (2015) Transcriptional regulation of the alpha-synuclein gene in human brain tissue. Neurosci Lett 599:140–145. doi:10.1016/j.neulet.2015.05.029
Clough RL, Stefanis L (2007) A novel pathway for transcriptional regulation of alpha-synuclein. FASEB J 21:596–607. doi:10.1096/fj.06-7111com
Clough RL, Dermentzaki G, Haritou M, Petsakou A, Stefanis L (2011) Regulation of alpha-synuclein expression in cultured cortical neurons. J Neurochem 117:275–285. doi:10.1111/j.1471-4159.2011.07199.x
Sterling L, Walter M, Ting D, Schule B (2014) Discovery of functional non-coding conserved regions in the alpha-synuclein gene locus. F1000Res 3:259. doi:10.12688/f1000research.3281.2
Doxakis E (2010) Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem 285:12726–12734. doi:10.1074/jbc.M109.086827
Minones-Moyano E, Porta S, Escaramis G, Rabionet R, Iraola S, Kagerbauer B, Espinosa-Parrilla Y, Ferrer I, Estivill X, Marti E (2011) MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet 20:3067–3078. doi:10.1093/hmg/ddr210
Villar-Menendez I, Porta S, Buira SP, Pereira-Veiga T, Diaz-Sanchez S, Albasanz JL, Ferrer I, Martin M, Barrachina M (2014) Increased striatal adenosine A2A receptor levels is an early event in Parkinson’s disease-related pathology and it is potentially regulated by miR-34b. Neurobiol Dis 69:206–214. doi:10.1016/j.nbd.2014.05.030
Kabaria S, Choi DC, Chaudhuri AD, Mouradian MM, Junn E (2015) Inhibition of miR-34b and miR-34c enhances alpha-synuclein expression in Parkinson’s disease. FEBS Lett 589:319–325. doi:10.1016/j.febslet.2014.12.014
Xu W, Tan L, Yu JT (2015) The link between the SNCA gene and parkinsonism. Neurobiol Aging 36:1505–1518. doi:10.1016/j.neurobiolaging.2014.10.042
Kalivendi SV, Yedlapudi D, Hillard CJ, Kalyanaraman B (2010) Oxidants induce alternative splicing of alpha-synuclein: implications for Parkinson’s disease. Free Radic Biol Med 48:377–383. doi:10.1016/j.freeradbiomed.2009.10.045
Beyer K, Domingo-Sabat M, Humbert J, Carrato C, Ferrer I, Ariza A (2008) Differential expression of alpha-synuclein, parkin, and synphilin-1 isoforms in Lewy body disease. Neurogenetics 9:163–172
Beyer K (2006) Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol 112:237–251
Lee HJ, Choi C, Lee SJ (2002) Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem 277:671–678
Beyer K, Lao JI, Carrato C, Mate JL, Lopez D, Ferrer I, Ariza A (2004) Differential expression of alpha-synuclein isoforms in dementia with Lewy bodies. Neuropathol Appl Neurobiol 30:601–607. doi:10.1111/j.1365-2990.2004.00572.x
Beyer K, Humbert J, Ferrer A, Lao JI, Carrato C, Lopez D, Ferrer I, Ariza A (2006) Low alpha-synuclein 126 mRNA levels in dementia with Lewy bodies and Alzheimer disease. Neuroreport 17:1327–1330. doi:10.1097/01.wnr.0000224773.66904.e7
Beyer K, Domingo-Sabat M, Lao JI, Carrato C, Ferrer I, Ariza A (2008) Identification and characterization of a new alpha-synuclein isoform and its role in Lewy body diseases. Neurogenetics 9:15–23
Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ, Unni VK (2011) Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci 31:14508–14520. doi:10.1523/JNEUROSCI.1560-11.2011
Ward WF (2002) Protein degradation in the aging organism. Prog Mol Subcell Biol 29:35–42
Xilouri M, Brekk OR, Stefanis L (2013) Alpha-synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol 47:537–551. doi:10.1007/s12035-012-8341-2
Xia Y, Rohan de Silva HA, Rosi BL, Yamaoka LH, Rimmler JB, Pericak-Vance MA, Roses AD, Chen X, Masliah E, DeTeresa R, Iwai A, Sundsmo M, Thomas RG, Hofstetter CR, Gregory E, Hansen LA, Katzman R, Thal LJ, Saitoh T (1996) Genetic studies in Alzheimer’s disease with an NACP/alpha-synuclein polymorphism. Ann Neurol 40:207–215
Touchman JW, Dehejia A, Chiba-Falek O, Cabin DE, Schwartz JR, Orrison BM, Polymeropoulos MH, Nussbaum RL (2001) Human and mouse alpha-synuclein genes: comparative genomic sequence analysis and identification of a novel gene regulatory element. Genome Res 11:78–86
Chiba-Falek O, Nussbaum RL (2001) Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum Mol Genet 10:3101–3109
Goldman SM, Umbach DM, Kamel F, Tanner CM (2015) Head injury, alpha-synuclein Rep1 and Parkinson’s disease: a meta-analytic view of gene-environment interaction. Eur J Neurol 22:e75. doi:10.1111/ene.12694
Kay DM, Factor SA, Samii A, Higgins DS, Griffith A, Roberts JW, Leis BC, Nutt JG, Montimurro JS, Keefe RG, Atkins AJ, Yearout D, Zabetian CP, Payami H (2008) Genetic association between alpha-synuclein and idiopathic Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet 147B:1222–1230
Farrer M, Maraganore DM, Lockhart P, Singleton A, Lesnick TG, de Andrade M, West A, de Silva R, Hardy J, Hernandez D (2001) Alpha-synuclein gene haplotypes are associated with Parkinson’s disease. Hum Mol Genet 10:1847–1851
Mizuta I, Nishimura M, Mizuta E, Yamasaki S, Ohta M, Kuno S (2002) Meta-analysis of alpha synuclein/NACP polymorphism in Parkinson’s disease in Japan. J Neurol Neurosurg Psychiatry 73:350
Mellick GD, Maraganore DM, Silburn PA (2005) Australian data and meta-analysis lend support for alpha-synuclein (NACP-Rep1) as a risk factor for Parkinson’s disease. Neurosci Lett 375:112–116
Cronin KD, Ge D, Manninger P, Linnertz C, Rossoshek A, Orrison BM, Bernard DJ, El-Agnaf OM, Schlossmacher MG, Nussbaum RL, Chiba-Falek O (2009) Expansion of the Parkinson disease-associated SNCA-Rep1 allele upregulates human alpha-synuclein in transgenic mouse brain. Hum Mol Genet 18:3274–3285. doi:10.1093/hmg/ddp265
Linnertz C, Saucier L, Ge D, Cronin KD, Burke JR, Browndyke JN, Hulette CM, Welsh-Bohmer KA, Chiba-Falek O (2009) Genetic regulation of alpha-synuclein mRNA expression in various human brain tissues. PLoS one 4:e7480. doi:10.1371/journal.pone.0007480
Beyer K, Humbert J, Ferrer A, Lao JI, Latorre P, Lopez D, Tolosa E, Ferrer I, Ariza A (2007) A variable poly-T sequence modulates alpha-synuclein isoform expression and is associated with aging. J Neurosci Res 85:1538–1546. doi:10.1002/jnr.21270
McCarthy JJ, Linnertz C, Saucier L, Burke JR, Hulette CM, Welsh-Bohmer KA, Chiba-Falek O (2011) The effect of SNCA 3′ region on the levels of SNCA-112 splicing variant. Neurogenetics 12:59–64. doi:10.1007/s10048-010-0263-4
Sotiriou S, Gibney G, Baxevanis AD, Nussbaum RL (2009) A single nucleotide polymorphism in the 3′UTR of the SNCA gene encoding alpha-synuclein is a new potential susceptibility locus for Parkinson disease. Neurosci Lett 461:196–201. doi:10.1016/j.neulet.2009.06.034
Kruger R, Vieira-Saecker AM, Kuhn W, Berg D, Muller T, Kuhnl N, Fuchs GA, Storch A, Hungs M, Woitalla D, Przuntek H, Epplen JT, Schols L, Riess O (1999) Increased susceptibility to sporadic Parkinson’s disease by a certain combined alpha-synuclein/apolipoprotein E genotype. Ann Neurol 45:611–617
McCulloch CC, Kay DM, Factor SA, Samii A, Nutt JG, Higgins DS, Griffith A, Roberts JW, Leis BC, Montimurro JS, Zabetian CP, Payami H (2008) Exploring gene-environment interactions in Parkinson’s disease. Hum Genet 123:257–265
Peuralinna T, Myllykangas L, Oinas M, Nalls MA, Keage HA, Isoviita VM, Valori M, Polvikoski T, Paetau A, Sulkava R, Ince PG, Zaccai J, Brayne C, Traynor BJ, Hardy J, Singleton AB, Tienari PJ (2015) Genome-wide association study of neocortical Lewy-related pathology. Ann Clin Transl Neurol 2:920–931. doi:10.1002/acn3.231
Kim WS, Kagedal K, Halliday GM (2014) Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Res Therapy 6:73. doi:10.1186/s13195-014-0073-2
Chen Y, Wei QQ, Ou R, Cao B, Chen X, Zhao B, Guo X, Yang Y, Chen K, Wu Y, Song W, Shang HF (2015) Genetic variants of SNCA are associated with susceptibility to Parkinson’s disease but not amyotrophic lateral sclerosis or multiple system atrophy in a Chinese population. PLoS one 10:e0133776. doi:10.1371/journal.pone.0133776
Ammal Kaidery N, Tarannum S, Thomas B (2013) Epigenetic landscape of Parkinson’s disease: emerging role in disease mechanisms and therapeutic modalities. Neurotherapeutics 10:698–708. doi:10.1007/s13311-013-0211-8
Jowaed A, Schmitt I, Kaut O, Wullner U (2010) Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci off J Soc Neurosci 30:6355–6359. doi:10.1523/JNEUROSCI.6119-09.2010
Matsumoto L, Takuma H, Tamaoka A, Kurisaki H, Date H, Tsuji S, Iwata A (2010) CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS one 5:e15522. doi:10.1371/journal.pone.0015522
Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, Adame A, Rockenstein E, Masliah E (2011) Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem 286:9031–9037. doi:10.1074/jbc.C110.212589
Song Y, Ding H, Yang J, Lin Q, Xue J, Zhang Y, Chan P, Cai Y (2014) Pyrosequencing analysis of SNCA methylation levels in leukocytes from Parkinson’s disease patients. Neurosci Lett 569:85–88. doi:10.1016/j.neulet.2014.03.076
Acknowledgments
We thank Dr. W.K. Gottschalk for the useful comments and discussions. This work was funded in part by the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS; R01 NS085011 to O.C.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tagliafierro, L., Chiba-Falek, O. Up-regulation of SNCA gene expression: implications to synucleinopathies. Neurogenetics 17, 145–157 (2016). https://doi.org/10.1007/s10048-016-0478-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-016-0478-0