
Abstract
In this study, the effects of protein adsorption on the electrochemical corrosion behavior of commercially pure niobium in phosphate-buffered saline (PBS) solutions with several concentrations (0–4 g L–1) of bovine serum albumin (BSA) were investigated via both electrochemical measurements and surface characterization. For low concentrations of BSA, the adsorbed protein molecules are insufficient to form a barrier layer, resulting in slight decrease in the cathodic reaction via the inhibition effect. Meanwhile, the anodic reaction is accelerated by the chelation effect between the protein molecule and metal ions. When the added BSA reaches saturation capacity, the adsorbed protein amount is approximately 1.8 μg cm–2, corresponding to approximately 1.2 adsorbed layers, facilitating the formation of a complete barrier layer. The inhibition effect of adsorbed protein molecules becomes more dominant for both cathodic and anodic reactions. The results of this study will help provide a better understanding of the mechanism of protein adsorption on the corrosion behavior of biometallic alloys.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Niobium (Nb) and its alloys have recently come under focus in the biomedical implant field, owing to their excellent corrosion resistance and associated biocompatibility [1, 2]. It is well-known that Nb very readily forms a stable and protective oxide layer which acts as a barrier layer between the metal matrix and its environment [3]. Several electrochemical studies have confirmed that benefiting from such a highly inert and unreactive oxide layer, Nb, and its alloys exhibited very low corrosion rates without pitting or transpassive behavior in various physiological scenarios [4, 5]. Because high corrosion resistance of metallic implants avoids the risks of ion release into the human body and possibly adverse biological reactions, superior biocompatibility of Nb and its alloys is assured [6].
After insertion into the human body, implant materials are immediately exposed to body fluid with not only inorganic ions (e.g., Na+, Cl–, K+, and HCO3–) but also organic compounds, especially various proteins [7, 8]. It is generally accepted that protein adsorption occurs rapidly after the interaction of implant materials with inorganic ions. Thus, protein adsorption would be likely to affect the corrosion behavior of implant materials besides the inevitable corrosion attack caused by inorganic anions. In the 1980s, Clark et al. [6] studied the corrosion behavior of several pure metals including Al, Co, Cu, Cr, Mo, Ni, and Ti in saline solution with albumin or fibrinogen, finding that the addition of protein had no significant influence on the corrosion of Ti and Al but presented an inhibition to Mo and a slight acceleration to Cr and Ni. It is worth noting that there is a significant increase, by a factor of 30–40, in the dissolution rate of Co and Cu. Karimi et al. [9, 10] investigated the effects of bovine serum albumin (BSA) addition on the corrosion behavior of 316 L, Co–28Cr–6Mo, and Ti–6Al–4V alloys in phosphate-buffered saline (PBS) solution, and also found that the effects varied with alloy type. The presence of BSA increased the passive film stability of the 316 L and Co–28Cr–6Mo alloys. However, BSA in the concentration range of 0.2–2 g L–1 reduced the passive film stability of the Ti–6Al–4V samples. Cheng et al. [11] found that the addition of 0.05 g L–1 BSA in PBS solution significantly moved the open-circuit potentials (OCP) toward a more negative (cathodic) potential and inhibited the cathodic corrosion reaction but did not significantly change the corrosion resistance at the OCP. Recently, Wang et al. [12] found that a competitive adsorption between PO43– and BSA could influence the corrosion behavior of Nb.
From the analyses of several mechanisms in previous studies, protein adsorption can affect the corrosion behavior of metals mainly though inhibition and/or chelation effects [6, 11]. Generally, protein molecules can form a barrier layer between metallic oxide film and human body environment, not only inhibiting oxygen diffusion but also suppressing the transport of positive metallic ions and corrosion products. Then, the charge-transfer process, which influences the growth and dissolution of the oxide film is inhibited. However, in another way, proteins have an isoelectric point of 4.7–5.2 and are negatively charged in blood serum [13, 14]. The interactions of metal cations with proteins generate colloidal organometallic complexes [15]. This chelation effect could significantly enhance the rate of metal dissolution and corrosion rate. Owing to the coupled inhibition and chelation effects of protein adsorption, the exact correlation between protein adsorption amount and corrosion behavior of Nb needs to be further elucidated.
Thus, considering recent studies, the effect of protein adsorption on the corrosion behavior of commercially pure (c.p.) Nb is investigated in this study. The protein adsorption behavior is characterized by time-of-flight secondary ion mass spectrometry (ToF-SIMS), and the adsorption amount is quantitatively obtained by the micro bicinchoninic acid (BCA) protein assay. The electrochemical corrosion behavior of c.p. Nb in PBS solutions with a wide range of BSA concentrations is investigated using various electrochemical methods. Moreover, with help of scanning electron microscopy (SEM), atomic force microscopy (AFM), and X-ray photoelectron spectroscopy (XPS), the effect of protein adsorption on the chemical nature of passive films and changes in the surface topography at the nanoscale are examined. The results of this study will help provide a better understanding of the mechanism of protein adsorption on the corrosion behavior of biometallic alloys.
Material and methods
Materials and test electrolyte
The wrought sheets of high purity type-1 Nb (UNS R04200, ASTM B393) were obtained from Baoji Chuangqi Metals Co. Ltd., China. The chemical composition and microstructure of c.p. Nb used in this work are provided in supplementary material. For electrochemical measurements, cubic metallic specimens were prepared with dimensions of 10 mm × 10 mm × 10 mm. They were embedded into a cold-cure epoxy resin, in which only one face was exposed to the electrolyte. The samples for immersion test were prepared with dimensions of 10 mm × 10 mm × 2 mm. All samples were mechanically ground with SiC emery paper up to 3000 grit, roughly polished using 1-μm diamond polishing paste, and finally fine-polished using a 0.04-μm silica suspension to reach a mirror-like surface finish. The arithmetic average roughness values, Ra, were 5 ± 1 nm and determined with an Olympus LEXT OLS4000 3D measuring laser confocal microscopy (LCM, Olympus, Japan) taken from surface areas of 40 μm × 40 μm. Prior to the tests, the specimens were ultrasonically cleaned with deionized water, acetone, and ethanol, then dried using compressed air.
Phosphate-buffer saline (PBS) solution was used to mimic the body fluid environment. This solution was chemically composed of NaCl (8 g L–1), KCl (0.2 g L–1), Na2HPO4·12H2O (2.9 g L–1), and KH2PO4 (0.2 g L–1), and prepared using deionized water with a resistivity of 18.2 MΩ cm and certified reagents supplied by Sinopharm Chemical Reagent Co., Ltd., China. The freshly prepared PBS solution contained a pH value of 7.4. Bovine serum albumin (BSA) was chosen as the model protein in this work. BSA possesses a similar physicochemical property as human serum albumin and exhibits high solubility and stability in simulated body fluid [16]. Six concentrations of BSA (Sigma-Aldrich, A1933), i.e., 0.2, 0.4, 0.6, 1, 2, and 4 g L–1, were added to the PBS solution. The prepared solutions are stored in a refrigerator for at least 24 h before tests. The pH values of test solutions are stabilized at 7.4 during the test.
Protein adsoprtion characterization
A TOF-SIMS Model V spectrometer (ION-TOF GmbH, Muenster, Germany) was used to characterize the proteins. The chamber was pumped continuously to maintain an ultra-high vacuum (UHV), in which the normal pressure was less than 5 × 10–9 mbar. A bi-cluster liquid metal ion gun generated bunched 1 pA Bi+/Bi3+-primary ion pulses at 30 keV with 1.25 ns duration. The scan area is of 500 × 500 μm2. The positive and negative ion spectra were acquired over a range of 0–200 m/z. Positive ion spectra was calibrated to CH3+, C2H3+, and C3H5+ peaks, and negative ion spectra were calibrated to CH– and OH– peaks before further analysis. Wassell et al. [17] suggested that a steady adsorption state can be reached within 50 min after introducing the protein molecules. Frateur et al. [18] further confirmed that the kinetics of BSA adsorption was fast by the means of in situ EQCM measurement, and that a steady state was reached about 10 min after introducing the protein. Thus, the TOF-SIMS characterization was conducted after immersion in solution for 1 h in this work.
The amounts of BSA adsorbed on the c.p. Nb surface were determined by the micro-BCA protein assay as described in Ref. [19]. After incubation in the BSA-containing solution for 1 h at 37 °C, the specimens were rinsed in deionized water to remove the unabsorbed BSA molecules. Subsequently, the specimens were immersed in 5% sodium dodecyl sulfate (SDS) for 24 h at 37 °C and ultrasonically cleaned in deionized water for 10 min. The amounts of BSA adsorbed on the Nb surface were determined according to their concentration in SDS solution, using a micro-BCA protein assay kit (Pierce Biotechnology, USA, #23235). The standard curve is provided by the Kit producer and shows a linear working range for BSA equals 0.5 to 20 μg mL–1. For each concentration of BSA, five repeated measures data were used to ensure reproducibility.
Electrochemical measurements
The electrochemical experimental set-up was a conventional three-electrode electrochemical cell with a platinum foil as a counter electrode and saturated calomel electrode (SCE) as the reference electrode. Electrochemical measurements were performed with a Zahner Pro electrochemical workstation (Zahner, GmbH, Germany). The temperature during testing was maintained at 37 ± 0.1 °C through a water bath.
Before each electrochemical measurement, a cathodic pretreatment at – 1.2 VSCE for 3 min was carried out in order to provide more reproducible initial surface conditions for the specimen and assure good reproducibility [20]. Measurements of time-dependent OCP were performed right after sample immersion in the test solution, lasting for 3 h. Potentiodynamic polarization measurements were performed, starting at – 1 VSCE and ending at 1.2 VSCE, with a scan rate of 0.333 mV s–1. Electrochemical impedance spectroscopy (EIS) measurements were carried out at the end of the OCP measurements, ranging from 105 Hz down to 10–3 Hz at 10 cycles per decade, with an AC amplitude of 10 mV. The impedance data were analyzed with the ZsimpWin 3.2 software package. All electrochemical measurements were performed five times to minimize measurement error and enhance the measurement precision.
Surface characterization
Because the corrosion attack of c.p. Nb is tiny after short-term immersion, the specimens for surface characterization were immersed in PBS solution with several BSA concentrations for 4 weeks. The immersion test was conducted in centrifuge tube with 50 ml of capacities. The tube was sealed to avoid water evaporation. All the centrifuge tubes were placed in a shelf and kept in an incubator at 37 °C throughout the whole period of immersion. Electrolytes were replaced every day to avoid protein denaturation. After immersion, the specimens were ultrasonically cleaned with acetone and ethanol, and subsequently immersed in ethanol before transferring to the vacuum chamber. The specimen surfaces were not exposed to air throughout the entire process to minimize the compositional changes of surface oxide by contamination from the atmosphere.
Surface chemistry was analyzed by X-ray photoelectron spectrometry (XPS). The XPS measurements were conducted in an ESCALAB 250xi surface analysis system (Thermo VG, USA) with a monochromatic Al-Kα X-ray source of 1486.6 eV. The take-off angle was fixed at 90°. The beam spot size was 500 μm × 500 μm. Survey spectrum scanning was performed from 0 to 1000 eV, and narrow-scan spectra were obtained with energy step size of 0.1 eV. The measured binding energies were calibrated referring to the C 1 s peak with a binding energy value of 284.6 eV. Standard-binding energies of Nb, O, and F were determined according to the NIST XPS database [21]. Deconvolution of the peaks was performed with XPS Peak 4.1 software using line-profile fitting with Gaussian/Lorentzian functions. The Nb 3d5/2-3d3/2 peaks were constrained using a peak area ratio of 1.5 and peak separation of approximately 2.7 eV. The full width at half maximum (FWHM) of all peaks was fixed in the range of 1.5–2.0 eV.
Surface morphology was observed using field emission scanning electron microscopy (FESEM, Hitachi SU8220, Japan) in secondary electron imaging mode with an accelerating voltage of 15 kV and working distance of 9 mm. A three-dimensional topography at the nanoscale was analyzed using atomic force microscopy (AFM, Dimension FastScan, Bruker, USA). AFM images were acquired in peak force tapping mode at a scan frequency of 1 Hz. The scanning area was set as 20 × 20 μm. The images were analyzed using the Nanoscope 9 software package.
Results
Protein adsorption
Protein adsorption on the surface was qualitatively characterized with highly sensitive ToF-SIMS. Figure 1 shows the typical ToF-SIMS spectra of c.p. Nb surface after immersion in PBS solution with 4.0 g L–1 BSA for 1 h. Thirteen nitrogen-containing characteristic peaks of BSA are observed in the positive spectra at m/z = 18, 30, 31, 44, 68, 70, 72, 74, 84, 86, 110, 120, and 136, corresponding to NH4+, CH4N+, CH5N+, C2H6N+, C4H6N+, C4H8N+, C4H10N+, C3H8NO+, C5H10N+, C5H12N+, C6H10N2+, C8H10N+, and C8H10NO+, respectively [22]. The existence of these fragment peaks provides the evidence of the adsorption of the BSA molecule on the surface of c.p. Nb, subjected to PBS solution with BSA. In addition, the ion peaks of Nb+, as well as those of Na+, K+, Cl–, and PO3– from the salt in PBS solution were also detectable on the outermost layer of Nb.

ToF-SIMS spectra of c.p. Nb surface after immersing in PBS solution with 4.0 g L–1 BSA for 1 h: a positive ion mass spectrum, b negative ion mass spectrum
With the micro-BCA assay, the mass per unit area mprotein of adsorbed protein on c.p. Nb was determined quantitatively for the samples immersed in PBS solution, with a wide range of BSA concentration from 0.2 to 4.0 g L–1 (Fig. 2). The adsorption isotherm shows a typical Langmuir-type isotherm, and a saturation value of approximately 1.8 μg cm–1 is confirmed. This indicates that when the added protein concentration reaches a certain level, the adsorption amount no longer increases with further increase in the protein concentration. Satoshi et al. [23] studied the adsorption of BSA on SiO2, TiO2, ZrO2, and Al2O3, and similar adsorption behavior was confirmed.

Adsorption amount and layers of BSA protein on the surface of c.p. Nb after immersing in PBS solution with several concentrations of BSA for 1 h
Based on the BSA adsorption amount, the number of layers for protein adsorption Lprotein can be estimated using following equations:
where M is the molecular weight of the protein, which is 66,460 g mol–1, and NA is Avogadro constant. Aprotein is the contact area between the molecule and specimen. Regarding the arrangement modes of protein molecule, two extreme situations need to be assumed. In the case one, the molecules of protein are supposed to be lined up vertically and densely close to the surface. The contact area is 2.7 × 2.7 nm2 [12]. The numbers of saturated adsorbed proteins layers are ~ 1.2 layers. In the case two, the molecules of protein are assumed to arrange horizontally, parallel to the surface of specimen, with the contact area is equal to 11.6 × 2.7 nm2 [12]. The numbers of saturated adsorbed proteins layers are ~ 5.2 layers. Since BSA protein molecules repel the other monomers electrostatically, BSA is known to form a single layer rather than multilayers [18, 24, 25]. Furthermore, Silva-Bermudez et al. [26] obtained the thickness of adsorbed BSA layers on NbOx by spectroscopic ellipsometry is around 10.3 nm which is close to the length of BSA protein molecules. Thus, the case one is more suitable for the practice. But it should be noted that the protein molecules are impossible to simply arrange as these two extreme cases, in a way either the vertical or the parallel to the alloy surface [27, 28]. This would result in the numbers of adsorbent layers larger than 1 by simplify calculating method. The calculated numbers of adsorbed protein layers are shown in Fig. 2. In the cases of 0.2–1 g L–1 BSA addition, the adsorbed protein layers increase which means the protein molecules not fully cover the c.p. Nb surface. Furthermore, when the BSA concentration is up to 1, 2, and 4 g L–1, the numbers of adsorbed proteins layers are stabilized at ~ 1.2, which means a complete coverage of adsorbed BSA. It is concluded that 1 g L–1 BSA is determined to be the threshold for one complete layer.
Open-circuit potential evolution
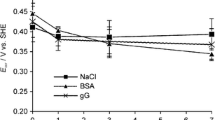
Figure 3a shows the evolution of the OCP for c.p. Nb in PBS solution with BSA of 0–4.0 g L–1 over 3 h. The OCP of c.p. Nb increases rapidly during the initial stage of immersion. With the extension of the immersion period, a near steady-state condition was approached as shown in the inset of Fig. 3a. Such features are typically attributed to the ongoing growth and stabilization of a passive film on the sample surface, which is similar to several typical self-passivation metals in PBS solution with or without BSA [9, 11, 29]. The steady-state OCP values, Eoc, determined at the end of OCP evolution are shown in Fig. 3b. It is apparent that the addition of 0.2 g L–1 BSA leads to decrease in OCP values as compared with the case of BSA0. As the BSA concentration increases from 0.2 to 1 g L–1, the OCP values continue to decrease. Meanwhile, when the BSA concertation further increases from 1 to 4 g L–1, the OCP values stabilized at ~ 715 mVSCE. Because steady-state OCP value is determined by a combination of electrode kinetics, as mixed potential theory indicates, the interpretation of such a trend of OCP versus BSA concentration is discussed below.

a Open-circuit potential evolution versus immersion period within 3 h for c.p. Nb in PBS solution with several concentrations of BSA and b plot of Eoc against BSA concentrations
Potentiodynamic polarization
Figure 4 shows the potentiodynamic polarization curves of c.p. Nb in PBS solution with a wide range of BSA concentrations. Basic electrochemical parameters were determined and summarized in Table 1.

Potentiodynamic polarization curves measured in PBS solution with several concentrations of BSA for c.p. Nb
As shown in the cathodic and anodic branches of polarization curves (Fig. 4), apparent Tafel regions which locate at the potential region from 250 to 100 mV below and above the corrosion potential, Ecorr, can be identified. Because the test solutions are weak alkaline and aerated, the oxygen reduction reaction (ORR) is predominant in the cathodic branch, namely [30],
The cathodic reaction Tafel slopes, –βc, are nearly identical at ~ 150 mV dec–1 in all cases, which indicates that no extra cathodic reactions besides ORR are involved after BSA addition. The cathodic reaction rate shows a reduction with BSA concentration, which becomes more apparent for BSA concentration exceeding 0.6 g L–1. The anodic reaction is as follows:
The anodic Tafel slopes, βa, are in the range of 80–90 mV dec–1 and slightly increase with BSA concertation.
The corrosion current density, jcorr, was obtained by Tafel linear extrapolation method. As shown in Fig. 5, for BSA concentration less than 0.6 g L–1, jcorr increases with BSA concentration. In the case of BSA0.6, maximum jcorr is 22 nA cm–2. Meanwhile, with further increase in the BSA concentration, jcorr exhibits a declining trend. A passivation plateau with nearly invariant current densities appears until the potential reaches 1.2 VSCE (not fully shown in Fig. 4). The passive current density, jpass, was taken at – 0.4 VSCE and is shown in Fig. 5. As the cathodic reaction rate is negligible in the passivation potential region, jpass is only determined by the anodic reaction rate. It is shown that with increasing BSA concentration up to 4.0 g L–1, jpass monotonically decreases, whereas for BSA concentration greater than 0.6 g L–1, the rising trend of jpass is lessened.

a OCP and Ecorr and bjcorr and jpass as a function of BSA concentration in PBS solution for c.p. Nb.
Electrochemical impedance spectroscopy
Figure 6a displays the Nyquist diagrams measured at OCP for c.p. Nb after immersion in PBS solution with several BSA concentrations for 1 h. Nyquist diagrams exhibit a characteristic single capacitive loop in the frequency range of 105 to 10–3 in all cases. Such impedance behavior is in accordance with several previous studies for c.p. Nb in simulated physiological environments [20]. As indicated, with an increase in BSA concentration from 0 to 0.6 g L–1, the capacitive loop shrinks accordingly. Meanwhile, further increase in BSA concentration from 0.6 to 4 g L–1 leads to the enlargement of the capacitive loop. The electrochemical parameters are fitted using the equivalent circuit (EC) model with one time constant as displayed in Fig. 6b. The paralleled capacitance and resistance, denoted as Qtotal and Rtotal, are attributed to the combination of protein adsorption layer, double layer, and passive films. Here, a constant phase element (CPE) was applied instead of pure capacitance, because the non-uniform current distribution and deviation induced by surface inconsistencies are taken into account in the CPE. The CPE is given as follows [31,32,33]:

a Nyquist diagrams, b equivalent circuit for c.p. Nb in PBS solution with several concentrations of BSA. The fitting curves are presented as solid lines together with the original data as symbols in the plots
where Q is the magnitude of the CPE, ω is the angular frequency, and n is the deviation parameter, which lies between 0.5 and 1. Calculated impedance properties in terms of the EC are summarized in Table 2. The plots of Rtotal and Qtotal versus BSA concentration are shown in Fig. 7. For BSA concentration less than 0.6 g L–1, Rtotal declines with increase in BSA concentration. Meanwhile, with further increase in the BSA concentration, Rtotal begins to increase. Such trend is reasonable since Rtotal is approximately inversely proportional to jcorr [5]. Qtotal exhibits an initial decrease and then levels off as the BSA concentration increases. Basically, the apparent surface capacitance can be attributed to two parts: double layer and passive film. As to passive film, with an assumption that the capacitance behaves as a parallel plate capacitor, there is a relation [12, 34],

Plot of Rtotal and Qtotal versus BSA concentration in PBS solution with several concentrations of BSA for c.p. Nb
where ε is the relative dielectric constant taken as 50 [35], ε0 is the permittivity of the vacuum (8.85 × 10–14 F cm–1), and A is the effective surface area [12]. Based on the passive film analysis in section “XPS analysis,” the passive film thickness first decreases and then increases. Thus, the film capacitance presents a first increases and then decreases trend The film capacitance values determined to be 7.02 μF cm–1, 7.76 μF cm–1, and 6.60 μF cm–1 in the case of 0, 0.6, and 4 g L–1 BSA. It can be deduced that the decrease of total capacitance is mainly attributed to the BSA adsorption, which decreases the effective dielectric constant of surface layer on c.p. Nb and increases the double layer thickness [12, 34, 36].
Corrosion morphology analysis
Along with electrochemistry characterization, surface characterization was also conducted to provide a direct observation of surface degradation using SEM and AFM. Because c.p. Nb exhibits excellent corrosion resistance, the samples for corrosion morphology analysis are immersed in PBS solution with several BSA concentrations for 4 weeks until the corrosion attack accumulates to a certain extent. Figure 8 shows the typical SEM images of c.p. Nb before and after immersion in PBS solution with BSA concentrations of 0, 0.6, and 4 g L–1. Figure 9 shows the corresponding AFM three-dimensional topography at a much smaller scale. It is shown that in the case of BSA0 (Fig. 8b), a small number of tiny corrosion pits are present on the surface as compared with the as-polished sample. Such pits are more visible in the three-dimensional topography (Fig. 9b). The surface roughness Ra is determined to be 19.4 nm according to the AFM result. In the case of 0.6 g L–1 BSA addition, more pits are observed on the surface (Figs. 8c and 9c). These pits are of diameter ~ 1 μm and maximum depth of ~ 63 nm, and Ra increases up to 26.2 nm. In the case of 4 g L–1 BSA addition (Figs. 8d and 9d), the surface is the least attacked, compared with the cases of BSA0 and BSA0.6. The surface is smoother, and the Ra is determined to be 13.3 nm. The sequence of corrosion attack extent is: BSA0.6 > BSA0 > BSA4.

SEM images of a as-polished and corroded surfaces for c.p. Nb after 4 weeks immersion in PBS solution with b 0 BSA, c 0.6 g L–1 BSA, and d 4.0 g L–1 BSA

AFM three-dimensional topography of a as polished and corroded surfaces for c.p. Nb after 4 weeks immersion in PBS solution with b 0 BSA, c 0.6 g L–1 BSA, and d 4.0 g L–1 BSA
XPS analysis
XPS analysis was performed to characterize the chemical composition and thickness of the passive film on the surface of c.p. Nb, subjected to immersion in PBS solution with 0, 0.6, and 4 g L–1 BSA for 4 weeks. Fig. 10 displays the XPS survey spectrum of the outermost surface of the c.p. Nb. As shown in Fig. 10, in the case of BSA0, the main peaks for C, O, and Nb are detected, while in the case of BSA0.6 and BSA4, additional peak of N is detected. Although the elements of Na, K, Cl, and P are present in the solution, they were not detected in the spectrum owing to their low concentration incorporated in the Nb surface.

XPS survey spectrum of c.p. Nb after 4 weeks immersion in PBS solution with 0, 0.6, and 4.0 g L–1 BSA
Figure 11 a–d illustrates high-resolution XPS deconvoluted spectra of Nb 3d, O 1s, N 1s, and C 1s for the outermost layer of immersed c.p. Nb. As shown in Fig. 11a, in all cases, three peak doublets are shown. A pair of metallic state peaks (Nb) appear at approximately 203 eV for Nb 3d5/2 and 205.7 eV for Nb 3d3/2. Another doublet is located at approximately 204.5 eV for Nb 3d5/2 and 207.2 eV for Nb 3d5/2, which corresponds to suboxides of Nb, such as NbO2 and NbO. The third doublet appears at approximately 208.4 eV for Nb 3d5/2 and 211.1 eV for Nb 3d5/2, which refers to full oxides of Nb, such as Nb2O5. The large amount of metallic Nb indicates that the oxide must be very thin [37, 38]. This allows the film thickness to be estimated from the Nb 3d spectra, assuming a bi-layer model of a thin Nb-oxide film on an infinite substrate [37, 39, 40]:

XPS high-resolution deconvoluted spectra of c.p. Nb after 4 weeks immersion in PBS solution with 0, 0.6, and 4.0 g L–1 BSA: a Nb 3d, b O 1s, c N 1s, and d C 1s
where d is the thickness of the passive film, θ is the take-off angle with respect to the surface, which is 90° in this study, λi is the inelastic mean free electron path, Фi is cation density, which can be calculated by dividing the physical density by the molecular weight, yielding \( {\varPhi}_{{\mathrm{Nb}}_2{\mathrm{O}}_5} \) = 0.0168 mol cm–3 and ΦNb = 0.0922 mol cm–3, and \( {\lambda}_{{\mathrm{Nb}}_2{\mathrm{O}}_5} \) and λNb are taken to be 3.43 nm and 1.93 nm, respectively [34]. By the calculations, the thickness of passive film formed on Nb in the case of BSA0 is determined to be 6.3 nm.
In the case of BSA0.6, the ratio of Nb5+ oxide to metallic Nb decreases as compared with the case of BSA0. Thus, based on Eq. 6, the passive film thickness is certainly thinner and is calculated to be 5.7 nm. In the case of BSA4, the ratio of Nb5+ oxide to metallic Nb is close to the case of BSA0, and the passive film thickness is calculated to be 6.7 nm. Typically, a thick passive film is resulted from an increased oxidation or oxide adjustment reactions, which happens in less corrosive solution [20].
As shown in Fig. 11b, the O 1s peaks decompose into two peaks at 529.9 eV and 531.6 eV, which are attributed to the oxygen in the metal–O(M–O) bond and to the O–H bond, respectively. It is shown that in the cases of BSA0.6 and BSA4, the ratio of [O–H]/[M–O] is higher than that in the case of BSA0, which may be attributed to the fact that more adsorbed protein results in the formation of more hydroxyl compounds and absorbed water. As shown in Fig. 11c, the N 1s signal is located at a binding energy of 399.8 eV in the cases of BSA0.6 and BSA4, which is attributed to the amino and amide groups from the BSA layer adsorbed on the metal surface. As shown in Fig. 11d, The C 1s peak includes the prominent hydrocarbon (C–H), hydroxyl carbon (C–O), carbon bound to nitrogen (C–N), and amide carbon (N–C=O). The corresponding peaks are located at the binding energies of 284.6, 286.0, and 287.8 eV, respectively. The signal with low binding energy for C–H bonds is attributed to air contamination during the sample transfer into the XPS chamber, while those with high binding energy are associated with the C–O, C–N, and N–C=O groups of protein molecules [7]. The spectra from elements C, N, and O suggest the presence of characteristic protein functional groups.
Discussion
In the scope of this study, both SIMS and XPS spectrum analysis show the characteristics of adsorbed protein molecules on the c.p. Nb surface (Fig. 1, Fig. 11c and d). Typically, the IEP value of Nb2O5 in various solutions is approximately 4 which indicates that the Nb surface is negatively charged in this work [41]. Since the BSA protein molecules are also negatively charged, the driving force for BSA adsorption is not electrostatic interactions. Yan et al. [42] concluded that hydrophobic interaction could govern the process of the protein adsorption on biometal when electrostatic interaction is absent. Furthermore, McCafferty [43] suggested that adsorption of negative charged substances onto the negatively charged oxide surface may proceed through the operation of van der Waals forces. BSA adsorption on the c.p. Nb surface in PBS solution exhibits a concentration-dependent behavior. When the protein adsorption is unsaturated, the amount of adsorption increases with BSA concentration. With the addition of 1 g L–1 BSA to the PBS solution, the saturation adsorption amount is reached. The saturation capacity of BSA adsorption is approximately 1.8 μg cm–2. Wassell et al. [17] studied the effect of protein concentrations on the BSA adsorption behavior on the Ti powders. It was revealed that the amount of BSA on Ti powder was saturated at a steady level of 1–2 μg cm–2. However, such adsorption capacity is higher than that estimated by EQCM measurements, which is 424 ng cm–2 on passivated chromium by Frateur et al. [18], 457 ng cm–2 on chromium metal by Lundin et al. [44]. Such differences may attribute to the different alloy types. Furthermore, approximately 1.2 protein layers are adsorbed on the Nb surface after reaching the saturation capacity. Burgos-Asperilla et al. [24] investigated the protein adsorption kinetics onto Ti crystals in NaH2PO4 + CaCl2 solution with 2.52 g L–1 BSA, and 1.5 protein layers were determined by quartz crystal microbalance (QCM). Silva-Bermudez et al. [26] studied BSA adsorption on films of stoichiometric Ta, Nb, Ti, and Zr oxides and 1.4–1.5 layers were determined by spectroscopic ellipsometry. For stainless steel and chromium metal, monolayer of adsorption BSA protein is also confirmed [14, 18, 44]. From above analyses, it is determined that 1 g L–1 BSA is the threshold to form a complete adsorption layer for Nb in PBS solution containing BSA.
A schematic diagram was drawn to visualize the effect of BSA adsorption on the corrosion behavior of c.p. Nb, as shown in Fig. 12. When the PBS solution is free of BSA, the basic events are the interaction of the c.p. Nb surface with inorganic ions. When low concentrations of BSA are added to the PBS solution, the adsorbed protein molecules are insufficient to form a barrier layer. However, the adsorbed protein molecules could reduce the cathodic reaction sites, which would result in a slight decrease in cathodic reaction rate [7, 10, 12]. This is consistent with the shift trend of cathodic branches in polarization curves (Fig. 4). In this case, the more notable influence is the chelation effect of protein molecules with metal ions. It has been well recognized that protein molecules can combine with metallic ions, migrating them from the passive film and electrolyte interface, and accelerate the process of metal dissolution [45]. By mixed potential theory, the values of Ecorr and OCP negatively shift. Besides, jcorr and jpass of BSA0.2 are both higher than those of BSA0, and as the BSA concentration increases to the saturation concentration (i.e., 0.6 g L–1), jcorr and jpass continue to increase. Meanwhile, Rp also exhibits an increase. Because of the very severe corrosion attack, more tiny corrosion pits are presented on the surface. The protective capacity of passive film is deteriorated, developing thinner thickness. Burstein et al. [46] studied the effects of bovine serum on the nucleation of corrosion pits in Ringer’s solution. They found that the presence of the organic components of the serum stimulates the nucleation of pits on 316 L stainless steel and commercially pure titanium.

Schematic diagram of corrosion behavior of c.p. Nb in PBS solution without and with low and high concentrations of BSA
For sufficiently high BSA concentration, the protein molecules form a complete adsorption layer. As mentioned earlier, the protein adsorption layer could be a barrier between the electrolyte and the oxide film. Thus, this layer decreases the transport rate of oxygen, corrosion products, and ions, and then impedes the charge-transfer process [47]. This occurs more notably with the cathodic reaction, which corresponds to the cathodic branch moving in the direction of smaller current density (Fig. 4). For the anodic dissolution, although more adsorbed protein molecules could introduce more chelation of protein molecules and metal ions, the transportation process of the chelate complex is inhibited. Thus, the anodic reaction rate no longer increases, which can be illustrated by the trend of jpass. In this case, the inhibition effect of protein adsorption outweighs the chelation effect. Furthermore, the cathodic reaction rate is more vital in determining the jcorr. It is shown that jcorr exhibits a decline as the BSA concentration increases from 0.6 to 4 g L–1. Correspondingly, Rtotal shows an increase with BSA concentration. The corrosion morphology confirmed that the corrosion attack is less severe in the case of high BSA concentration than for low BSA concentration. Moreover, the passive film is thicker than those of the BSA free and low BSA concentration cases.
Conclusions
By investigating the electrochemical behavior and surface characteristics, the effect of BSA adsorption on the corrosion behavior of c.p. Nb in PBS solution with several concentrations of BSA was examined. Within the limitations of this work, the main issues can be summarized as follows:
-
1.
BSA adsorption on c.p. Nb surface in PBS solution exhibits a concentration-dependent behavior. When the protein adsorption is unsaturated, the amount of adsorption increases with BSA concentration. One gram per square liter BSA is the threshold to reach the saturation adsorption amount and form a complete adsorption layer.
-
2.
For a low concentration of BSA added to the PBS solution, the adsorbed protein molecules result in a slight decrease in cathodic reaction. The more notable influence is the chelation effect of the protein molecule and metal ions, which results in the acceleration of the anodic reaction. The passive film is deteriorated, and more severe corrosion attack is present on the surface.
-
3.
For sufficiently high BSA concentration, the protein molecules can form a complete adsorption layer. The inhibition effect is more pronounced, especially for the cathodic reaction, as the cathodic branch moves to a much smaller current density. Chelation effect is insignificant for accelerating anodic reaction rate. The corrosion morphology confirmed that the corrosion attack is less severe in the case of high BSA concentration than in the case of low BSA concentration, whereas the passive film is thicker than those of the BSA free and low BSA concentration cases.
References
Matsuno H, Yokoyama A, Watari F, Uo M, Kawasaki T (2001) Biocompatibility and osteogenesis of refractory metal implants, titanium, hafnium, niobium, tantalum and rhenium. Biomaterials 22(11):1253–1262
O’Brien B (2015) Niobium biomaterials. In: Advances in Metallic Biomaterials. Springer, pp 245-272
Olsson C-OA, Vergé M-G, Landolt D (2004) EQCM study of anodic film growth on valve metals. J Electrochem Soc 151(12):B652–B660
Guan L, Li Y, Wang G, Zhang Y, Zhang L-C (2018) pH dependent passivation behavior of niobium in acid fluoride-containing solutions. Electrochim Acta 285:172–184
Wang W, Alfantazi A (2013) Effect of microstructure and temperature on electrochemical behavior of niobium in phosphate-buffered saline solutions. J Electrochem Soc 160(1):C1–C11
Clark GCF, Williams DF (1982) The effects of proteins on metallic corrosion. J Biomed Mater Res 16(2):125–134
Munoz AI, Mischler S (2007) Interactive effects of albumin and phosphate ions on the corrosion of CoCrMo implant alloy. J Electrochem Soc 154(10):C562–C570
Nie M, Wang CT, Qu M, Gao N, Wharton JA, Langdon TG (2014) The corrosion behaviour of commercial purity titanium processed by high-pressure torsion. J Mater Sci 49(7):2824–2831
Karimi S, Nickchi T, Alfantazi A (2011) Effects of bovine serum albumin on the corrosion behaviour of AISI 316L, Co–28Cr–6Mo, and Ti–6Al–4V alloys in phosphate buffered saline solutions. Corros Sci 53(10):3262–3272
Karimi S, Nickchi T, Alfantazi AM (2012) Long-term corrosion investigation of AISI 316L, Co–28Cr–6Mo, and Ti–6Al–4V alloys in simulated body solutions. Appl Surf Sci 258(16):6087–6096
Cheng X, Roscoe SG (2005) Corrosion behavior of titanium in the presence of calcium phosphate and serum proteins. Biomaterials 26(35):7350–7356
Wang W, Mohammadi F, Alfantazi A (2012) Corrosion behaviour of niobium in phosphate buffered saline solutions with different concentrations of bovine serum albumin. Corros Sci 57:11–21
Bowen WR, Hughes DT (1990) Properties of microfiltration membranes. Part 2. Adsorption of bovine serum albumin at aluminium oxide membranes. J Membr Sci 51(1):189–200
Hedberg Y, Wang X, Hedberg J, Lundin M, Blomberg E, Odnevall Wallinder I (2013) Surface-protein interactions on different stainless steel grades: effects of protein adsorption, surface changes and metal release. J Mater Sci-Mater M 24(4):1015–1033
Woodman JL, Black J, Jiminez SA (1984) Isolation of serum protein organometallic corrosion products from 316LSS and HS-21 in vitro and in vivo. J Biomed Mater Res 18(1):99–114
Peters T Jr (1995) All about albumin: biochemistry, genetics, and medical applications. Academic press, Cambridge
Wassell DTH, Embery G (1996) Adsorption of bovine serum albumin on to titanium powder. Biomaterials 17(9):859–864
Frateur I, Lecoeur J, Zanna S, Olsson COA, Landolt D, Marcus P (2007) Adsorption of BSA on passivated chromium studied by a flow-cell EQCM and XPS. Electrochim Acta 52(27):7660–7669
Li X-M, Li H-Z, Wang S-P, Huang H-M, Huang H-H, Ai H-J, Xu J (2014) MRI-compatible Nb–60Ta–2Zr alloy used for vascular stents: haemocompatibility and its correlation with protein adsorption. Mater Sci Eng C 42:385–395
Li Y, Xu J (2017) Is niobium more corrosion-resistant than commercially pure titanium in fluoride-containing artificial saliva? Electrochim Acta 233:151–166
Wagner CD, Naumkin AV, Kraut-Vass A, Allison JW, Powell CJ, Rumble JR Jr (2003) NIST standard reference database 20. NIST XPS Database Version 3:251–252
Poleunis C, Rubio C, Compère C, Bertrand P (2002) Role of salts on the BSA adsorption on stainless steel in aqueous solutions. II. ToF-SIMS spectral and chemical mapping study. Surf Interface Anal 34(1):55–58
Fukuzaki S, Urano H, Nagata K (1996) Adsorption of bovine serum albumin onto metal oxide surfaces. J Ferment Bioeng 81(2):163–167
Burgos-Asperilla L, García-Alonso MC, Escudero ML, Alonso C (2010) Study of the interaction of inorganic and organic compounds of cell culture medium with a Ti surface. Acta Biomater 6(2):652–661
Shouren G, Kojio K, Takahara A, Kajiyama T (1998) Bovine serum albumin adsorption onto immobilized organotrichlorosilane surface: influence of the phase separation on protein adsorption patterns. J Biomat Sci-Polym E 9(2):131–150
Silva-Bermudez P, Rodil SE, Muhl S (2011) Albumin adsorption on oxide thin films studied by spectroscopic ellipsometry. Appl Surf Sci 258(5):1711–1718
Berlind T, Tengvall P, Hultman L, Arwin H (2011) Protein adsorption on thin films of carbon and carbon nitride monitored with in situ ellipsometry. Acta Biomater 7(3):1369–1378
Höök F, Vörös J, Rodahl M, Kurrat R, Böni P, Ramsden JJ, Textor M, Spencer ND, Tengvall P, Gold J, Kasemo B (2002) A comparative study of protein adsorption on titanium oxide surfaces using in situ ellipsometry, optical waveguide lightmode spectroscopy, and quartz crystal microbalance/dissipation. Colloids Surf, B 24(2):155–170
Aliofkhazraei M, Rouhaghdam AS, Sabouri M (2008) Effect of frequency and duty cycle on corrosion behavior of pulsed nanocrystalline plasma electrolytic carbonitrided CP-Ti. J Mater Sci 43(5):1624–1629
Flitt HJ, Schweinsberg DP (2005) A guide to polarisation curve interpretation: deconstruction of experimental curves typical of the Fe/H2O/H+/O2 corrosion system. Corros Sci 47(9):2125–2156
Xu Y, Li J, Qi M, Gu J, Zhang Y (2020) Effect of extrusion on the microstructure and corrosion behaviors of biodegradable Mg–Zn–Y–Gd–Zr alloy. J Mater Sci 55(3):1231–1245
Zhang J-W, Kong D-S, Zhang H, Du D-D, Wang N, Feng Y-Y, Li W-J (2015) A faradaic impedance study on the kinetic properties of water photosplitting at illuminated TiO2/solution interface. J Solid State Electrochem 19(11):3411–3423
Mandula TR, Srinivasan R (2017) Electrochemical impedance spectroscopic studies on niobium anodic dissolution in HF. J Solid State Electrochem 21(11):3155–3167
Milošev I, Kosec T, Strehblow HH (2008) XPS and EIS study of the passive film formed on orthopaedic Ti–6Al–7Nb alloy in Hank’s physiological solution. Electrochim Acta 53(9):3547–3558
Di Franco F, Zaffora A, Santamaria M (2018) Band gap narrowing and dielectric constant enhancement of (NbxTa(1-x))2O5 by electrochemical nitrogen doping. Electrochim Acta 265:326–335
McCafferty E (2010) Kinetics of corrosion. In: McCafferty E (ed) Introduction to Corrosion Science. Springer New York, New York, pp 146–159
Zhang L, Macdonald DD (1998) Segregation of alloying elements in passive systems—I. XPS studies on the Ni–W system. Electrochim Acta 43(18):2661–2671
Asami K, Hashimoto K (1987) X-ray photoelectron spectroscopy for corrosion studies. Langmuir 3(6):897–904
Bojinov M, Stancheva M (2015) Coupling between dissolution and passivation revisited – kinetic parameters of anodic oxidation of titanium alloys in a fluoride-containing electrolyte. J Electroanal Chem 737:150–161
Vickerman JC, Gilmore IS (2009) Surface analysis: the principal techniques, vol 2. Wiley Online Library,
Kosmulski M (2009) Surface charging and points of zero charge, vol 145. CRC Press, Boca Raton
Yan Y, Yang H, Su Y, Qiao L (2015) Albumin adsorption on CoCrMo alloy surfaces. Sci Rep 5:18403
McCafferty E (2003) Lewis acid/Lewis base effects in corrosion and polymer adhesion at aluminum surfaces. J Electrochem Soc 150(7):B342–B347
Lundin M, Hedberg Y, Jiang T, Herting G, Wang X, Thormann E, Blomberg E, Wallinder IO (2012) Adsorption and protein-induced metal release from chromium metal and stainless steel. J Colloid Interface Sci 366(1):155–164
Lewis AC, Kilburn MR, Heard PJ, Scott TB, Hallam KR, Allen GC, Learmonth ID (2006) The entrapment of corrosion products from CoCr implant alloys in the deposits of calcium phosphate: a comparison of serum, synovial fluid, albumin, EDTA, and water. J Orthop Res 24(8):1587–1596
Burstein G, Liu C (2007) Nucleation of corrosion pits in Ringer’s solution containing bovine serum. Corros Sci 49(11):4296–4306
Williams RL, Brown SA, Merritt K (1988) Electrochemical studies on the influence of proteins on the corrosion of implant alloys. Biomaterials 9(2):181–186
Funding
This work was funded by and the Guangdong Academic of Science' Project of Science and Technology Development (2018GDASCX-0114), the Guangzhou Foreign Science and Technology Cooperation Project (201907010004), and the National Natural Science Foundation of China (51701047).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors have declared that no conflict of interest exists.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 1337 kb)
Rights and permissions
About this article

Cite this article
Li, Y., Shi, C., Guan, L. et al. Correlation between protein adsorption and electrochemical corrosion behavior of niobium for bio-implant application. J Solid State Electrochem 24, 1325–1336 (2020). https://doi.org/10.1007/s10008-020-04634-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-020-04634-x