
Abstract
Redox mechanisms in which the redox transformation is coupled to other chemical reactions are of significant interest since they are regarded as relevant models for many physiological systems. Protein-film voltammetry, based on surface confined electrochemical processes, is a methodology of exceptional importance, which is designed to provide information on enzyme redox chemistry. In this work, we address some theoretical aspects of surface confined electrode mechanisms under conditions of square-wave voltammetry (SWV). Attention is paid to a collection of specific voltammetric features of a surface electrode reaction coupled with a follow-up (ECrev), preceding (CrevE) and regenerative (EC’) chemical reaction. While presenting a collection of numerically calculated square-wave voltammograms, several intriguing and simple features enabling kinetic characterization of studied mechanisms in time-independent experiments (i.e., voltammetric experiments at a constant scan rate) are addressed. The aim of the work is to help in designing a suitable experimental set-up for studying surface electrode processes, as well as to provide a means for determination of kinetic and/or thermodynamic parameters of both electrode and chemical steps.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the past three decades, square-wave voltammetry (SWV) emerged as a leading technique in the family of voltammetric methods. Its unique features stem from the specific shape of the applied exciting signal and the current sampling procedure that efficiently discriminates faradaic against charging current [1,2,3,4,5]. Hence, SWV is a highly sensitive voltammetric technique capable to detect miscellaneous analytes at nanomolar concentration level [6, 7]. Due to the complexity of the potential modulation applied, however, SWV is seldom explored as a technique for mechanistic evaluations. In spite of the fact that numerous theoretical models for various electrode mechanisms are presented so far, a wide application of SWV for kinetic and mechanistic studies is still a challenge. This holds true even for some electrode mechanisms considered to be “simple”. For instance, the so-called simple surface confined electrode reaction under conditions of square-wave voltammetry is attributed with rather complex voltammetric pattern [1, 6,7,8,9,10].
Enzymatic electrode reactions in the so-called protein film voltammetry [11,12,13,14,15,16,17] are probably the most important examples in the family of surface confined electrode processes. Indeed, electrode mechanisms of uniformly adsorbed redox active biological molecules coupled with follow-up or preceding chemical reactions attract significant attention as they are considered as adequate models for some physiological systems [7, 13, 16, 17]. We have presented recently several theoretical models of surface processes coupled to chemical reactions [18,19,20,21,22,23,24,25,26,27,28], revealing a set of intriguing, unique voltammetric features under conditions of SWV. In the present work, a further attempt is made to throw a light to the specific features of surface electrode processes coupled with a follow-up, preceding or regenerative chemical reaction. The nomenclature of these systems is surface ECrev, CrevE and EC′ (or surface catalytic mechanism), respectively. The aim of this cumulative work is to help experimentalists to characterize qualitatively particular electrode mechanism and to design a suitable experimental approach for estimation of kinetic and thermodynamic parameters governing surface electrode processes.
Mathematical models
We consider theoretically four electrode mechanisms adequate to the redox chemistry of firmly adsorbed enzymes in protein-film voltammetry:
-
1.
Simple surface confined electrode reaction (E)
-
2.
Surface confined catalytic (regenerative) electrode mechanism (surface EC′)
-
3.
Surface confined electrode reaction coupled with a follow-up reversible chemical reaction (surface ECrev)
-
4.
Surface confined electrode reaction coupled with a preceding reversible chemical reaction (surface CrevE)
In all mathematical models, we assume that all species are firmly immobilized on the electrode surface and the mass transfer is neglected. In addition, it is considered that the immobilized species form a uniform monolayer free of lateral interactions. In the schemes (2–4), the symbol “Y” is assigned to an electrochemically inactive compound that is present in a large excess. Consequently, the concentration of Y is virtually constant at the electrode surface in the course of voltammetry, and all chemical steps in reaction mechanisms (2–4) are considered to be of pseudo-first order. The solution of simple surface electrode mechanism (1) under SW voltammetric conditions can be found elsewhere [1, 7, 8, 17], while the corresponding solutions of the surface EC′, ECrev and surface CrevE mechanisms are reported in [1, 17,18,19, 26], respectively. The recurrent formulas for calculating dimensionless currents Ψ of theoretical SW voltammograms as a function of applied potential are given with Eqs. (5–8):
(5) is the recurrent formula for mechanism (1) i.e., simple surface redox reaction.
(6) is the recurrent formula for mechanism (2) (surface EC′).
(7) is the recurrent formula for mechanism (3) (surface ECrev).
(8) is the recurrent formula for mechanism (4) (surface CrevE).
In the recurrent formulas (I–IV), the reduction current is considered to be positive and the dimensionless current is defined as Ψ = I/[(nFSfΓ*)]. In the last equation, I is electric current, n is stoichiometric number of electrons, S is active electrode surface area, f is SW frequency (f = 1/2tp, where tp is the duration of a single potential pulse in SWV), and Γ*is the total surface concentration (i.e., the initial surface concentration of Ox species). Φ refers to the dimensionless potential (\( \varPhi =\frac{nF}{RT}\left(E-{E}^{\phi \hbox{'}}\right) \), whereEϕ’ is the formal redox potential, α is the electron transfer coefficient, while other symbols have their common meaning.
The dimensionless electrode kinetic parameter K = kso/f relates the formal rate constant kso to the SWV frequency. For the surface ECrev and CrevE mechanisms, Kchem = ε/f is the dimensionless chemical kinetic parameter, where ε = (kf + kb) is the sum of the first-order rate constant kf and kb of the forward and backward chemical reactions, respectively. In addition, the equilibrium constant is defined as Keq = kf/kb. For the surface EC′ catalytic mechanism, the dimensionless chemical kinetic parameter is defined as Kchem = kc/f, where kc is the first-order rate constant of the regenerative (catalytic) chemical reaction.
In addition, M is numerical integration factor defined as M = exp.[Κchem(m/50)] − exp.[Κchem(m-1)/50], where m is the serial number of the time intervals. If not otherwise stated, the parameters of applied potential were SW frequency f = 10 Hz, SW amplitude Esw = 50 mV and potential step dE = 4 mV and starting potential of 0.3 V. More details of the algorithms used in all models can be found in [1, 7, 17, 24, 29]. Commercially available software MATHCAD 14 was used for calculating all theoretical SW voltammograms reported in this work.
Results and discussions
Square-wave voltammetry is attributed with a unique shape of the potential modulation (see Scheme 1) that makes this technique very intriguing from several aspects.

Schematic representation of a potential cycle in SWV consisting of two adjacent oppositely oriented potential pulses. The current is measured at the end of potential pulses (in the sectors assigned with “If” and “Ib”). In the so-called dead time, the current is not measured, but the electrode reactions go on also in this time segment
Square-wave amplitude Esw (see Scheme 1) is actually the height of the potential pulse, i.e., a potential jump up and down (from several millivolts to hundred millivolts) at every single step of the underlying staircase potential. Consequently, SWV enables inspection of both oxidation and reduction processes at every single potential of the staircase ramp. The overall voltammetric response consists of three current components, i.e. forward (If), backward (Ib) and differential net component (Inet = If − Ib). The latter enables inspecting independently three current components as a function of relevant parameters of a certain electrochemical experiment. The current measuring in SWV takes place in a narrow time segment at the end of each potential pulse (If and Ib in the Scheme 1). In this way, one discriminates significantly faradaic from the charging current. Of course, the surface concentration variation of redox species prior to the current sampling in the so-called dead time (see scheme 1) plays an important role in overall features of SW voltammograms [25, 26]. Before we start elaborating some relevant characteristics of surface electrode processes coupled with chemical reactions, we shortly recall to the most important features of the simple surface mechanism (1), considered as a referent system in the context of the current study.
Simple surface confined electrode mechanism
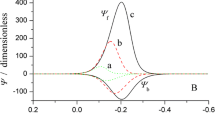
Shown in Fig. 1 is a series of theoretical square-wave voltammograms of a simple surface reaction, simulated for several values of the dimensionless electrode kinetic parameter K. As the rate of the electrode reaction increases, one observes significant enlargement of all current components. Beyond the maximum peak current reached for K ≈ 0.5 (Fig. 1b), one observes a considerable current decrease by further increasing of K (Fig. 1c–f). At the same time, cathodic (forward) peak starts to shift towards more positive potential values, while anodic (backward) peak shifts in opposite direction. As a consequence, splitting of the net SWV peak appears (Fig. 1d). The potential separation between the split net peaks increases in proportion to K (Figs.1d–f and 2b).

Simple surface redox reaction: Effect of the dimensionless electrode kinetic parameter K to the features of calculated square-wave voltammograms. The simulation parameters were SW frequency f = 10 Hz, SW amplitude Esw = 50 mV, potential step dE = 4 mV, temperature T = 298 K. In all simulations, the value of electron transfer coefficient was α = 0.5, while the number of electron exchanged was n = 2. The values of K are given in the charts. In all simulated voltammograms in this work, the reduction (forward) current is assigned with blue line, the oxidation (backward) current is assigned with red line, while the black line represents the “net” current

Simple surface redox reaction. a Quasi-reversible maxima of simple surface redox reactions simulated for three different values of SW amplitude. b Dependence of the potential separation of “split net SWV peaks” as a function of log(K), simulated for square-wave amplitudes of 30 mV (1), 40 mV (2) and 50 mV (3). All other simulation parameters are same as those in Fig. 1
Shown in Fig. 2a is the relationship of net peak current as a function of log(K), simulated for three different values of the SW amplitude. These so-called quasi-reversible maxima imply that the maximal net peak current is a function of the electrode reaction rate (via K), as well as the SW amplitude. This is to be expected, since the kinetics of the electrode reaction depends not only on the formal rate constant of the electron transfer (and on temperature) but also on the potential applied, which is determined by the staircase potential and the pulse applied (i.e., SW amplitude).
The phenomenon of quasi-reversible maximum emerges when the rate of electron transfer and the duration of the potential pulses in SWV are synchronized [1, 6,7,8,9,10].
On the other hand, the splitting of the net peak is attributed to electrode processes with rather fast electron transfer kinetics [1, 7, 8, 17]. When the electron transfer rate is large, the time required for redox transformation of Ox(ads) to Red(ads) at the potential of a given pulse becomes very short. Hence, when the pulse duration is longer than the time needed to achieve the redox transformation of Ox(ads) to Red(ads), the measured current at the end of the pulse is very small, resulting in a minute overall voltammetric response (Fig. 1e, f). Both “quasi-reversible maximum” and “net SWV peak splitting” can be explored to get an access to the formal rate constant value of a studied system in a very elegant way. A detailed overview of the corresponding methods can be found elsewhere [1, 6,7,8,9,10, 17]. In [27, 28, 30,31,32,33,34,35,36,37,38,39], one can find a detailed description how to alternatively evaluate kso and the electron transfer coefficient α.
Surface confined catalytic (regenerative) electrode mechanism (surface EC′):
In the family of surface electrode processes coupled with chemical reactions, the simplest case is probably the surface catalytic mechanism (surface EC′) (2). Here, an irreversible chemical redox reaction between electrochemically generated product Red(ads) with a given reactant Y (present in a large excess in the voltammetric cell) leads to regeneration of the initial reactant Ox(ads). Shown in Fig. 3 are SW voltammograms simulated for moderate rate of electrode reaction (K = 0.5) and several values of the catalytic (chemical) parameter Kchem. An increase of the reduction (forward) and a decrease of the oxidation (backward) current branch with increasing of Kchem is a typical feature for this mechanism. In parallel, the phenomenon is accompanied by an increase of the net peak current. As the catalytic parameter Kchem gets larger, both reduction and oxidation current gain identical sign and approach to each other with their magnitudes. It happens when the rate of the catalytic reaction is much higher than the rate of the redox transformation at the working electrode surface. As a consequence, a multiple reuse of the starting electroactive material occurs in the time-measuring segment of each potential pulse, which produces current in proportion to Kchem. Contrary to cyclic voltammetry, where one observes a steady-state sigmoid-shaped cyclic voltammograms for large rates of the regenerative reaction [2, 3, 20, 40, 41], in SWV, one finds a well-defined net SW peak for any value of the catalytic parameter Kchem (Fig. 3c, d). This effect is a consequence of the differential nature of the net SW component. In addition, it stems from the features of the forward and backward current components, which are always separated to some extent at the potential axis, if SW amplitudes larger than 10 mV are applied [22].

Surface EC′ catalytic mechanism: case with moderate electron transfer rate: Effect of the chemical (catalytic) parameter Kchem to the features of calculated SW voltammograms. The values of the catalytic parameter Kchem were 0.0001 (a), 0.316 (b), 1 (c) and 10 (d). The value of the dimensionless electrode kinetic parameter K was 0.5. All other simulation parameters were same as those in Fig. 1
For Kchem > 1, a regular shift of the net SWV peak potentials in a negative direction is detected (Fig. 3d). Indeed, this is because more energy is needed to reduce the new influx of Ox(ads) species resupplied via chemical redox reaction of Red(ads) with Y. This is clearly presented in Fig. 4a, where only the net SWV peaks are shown, simulated for several high values of the catalytic parameter Kchem. In Fig. 4b, the forward and backward current components of the corresponding net voltammograms of Fig. 4a are presented. Obviously, both components gain a sigmoid shape as typical for steady-state processes. However, both currents do not merge to each other, as in cyclic voltammetry [2, 40, 41], even for large values of the chemical parameter Kchem. The reason for this behaviour is in the influence of applied square-wave amplitude applied, as it is explained in more details in [22].

Surface EC′ catalytic mechanism: case with moderate electron transfer rate: Effect of the chemical (catalytic) parameter Kchem to the features of calculated net SW voltammograms (a) and to the corresponding reduction and oxidation SW currents (b). In both cases, the values of the catalytic parameter Kchem were 0.001 (1), 2.0 (2), 3.16 (3), 5.0 (4) and 10 (5). b The value of the dimensionless electrode kinetic parameter K was 0.10. All other simulation parameters were same as in Fig. 1
If the electrode reaction is very fast, then the splitting of the net SWV peak occurs (Fig. 5a). Increasing the rate of the catalytic reaction leads to a decrease of the backward (oxidation) current component and corresponding increase of the forward (reduction) current (Fig. 5b, c). Eventually, for Kchem > 0.02, the splitting vanishes and a single net peak is observed (Fig. 5d). This is a unique feature, which can serve as a criterion for qualitative characterization of this mechanism [18, 20].

Surface EC′ catalytic mechanism: case with fast electron transfer rate: Effect of the chemical (catalytic) parameter Kchem to the features of calculated SW voltammograms. The values of the catalytic parameter Kchem were 0.00001 (a), 0.001 (b), 0.01 (c) and 0.1 (d). The value of the dimensionless electrode kinetic parameter K was 10. All other simulation parameters were same as those in Fig. 1
In [22], we demonstrated that in the region of moderate-to-large rate of the regenerative reaction (roughly −1.5 < log(Kchem) < 1.5), the net peak potential Enet,p is a linear function of log(Kchem) (Fig. 6a).

Surface EC′ catalytic mechanism: case with slow and moderate electron transfer rate. a Dependence of the net SWV peak potentials as a function of logarithm of catalytic parameter log(Kchem). The curves are simulated for three values of dimensionless electrode kinetic parameter, i.e. log(K) 0f − 0.40 (1), − 1.0 (2) and − 2.0 (3). b Dependence of the limiting currents of SW voltammograms (measured at potentials of − 0.30 V) from Fig. 4b as a function of catalytic parameter Kchem, simulated for K of 0.10. All other simulation parameters were same as those in Fig. 1
While the slope of the line Ep vs. log(Kchem) is − 59 mV, the intercept is defined as follows: intercept = EoOx(ads)/Red(ads) + 2.303 RT(αnF)−1 log(K). If the value of electron transfer coefficient α is previously known [1, 30], the latter equation allows estimation of the electrode kinetic parameter K (hence the formal rate constant kso, since K = kso /f) by varying the concentration of the regenerative agent Y. This comes from the fact that the dimensionless chemical parameter is defined as Kchem = kc/f, where kc = kc′c(Y) and kc′ is the real second-order rate constant of the regenerative reaction. Thus, the rate of the surface electrode reaction can be estimated in a time-independent experiment, i.e. at constant SW frequency.
Furthermore, from the limiting current of the steady-state forward and backward currents (Fig. 4b), the rate of the regenerative chemical reaction can be estimated in a simple way. Shown in Fig. 6b is the dependence of the limiting current of both components as a function of the chemical parameter Kchem. The slope of the linear function allows determination of kc′ only by altering the concentration of regenerative agent taking into account that Kchem = kc′c(Y).
Considering features of SW voltammograms presented in Figs. 4 and 5, and the dependences presented in Fig. 6, it follows that the kinetics of both electrode and chemical reaction is accessible in a time-independent experiment. Thus, it can be achieved at constant scan rate and frequency of the SW voltammetric experiment, taking advantage of altering the concentration of regenerative agent Y only.
Surface-confined electrode reaction coupled with a follow-up reversible chemical reaction (surface ECrev)
The surface ECrev mechanism is often encountered in the voltammetry of various immobilized enzymatic reactions, as well as with many lipophilic drugs prone to adsorb at the electrode surface [1, 6, 7, 10,11,12,13,14,15,16,17, 40,41,42,43,44,45,46,47,48,49]. We consider here a surface confined electrode reaction in which the electrochemically generated redox product is involved in a reversible follow-up chemical reaction [26]. The analysis is focused mainly on the ECrev mechanism featuring fast electron transfer.
Voltammetric features are mainly affected by the electrode kinetic parameter K = kso/f, as in previous mechanisms, as well as by the equilibrium constant of the chemical step Keq = kf/kb and the overall rate of the follow-up chemical reaction via the dimensionless chemical kinetic parameter Kchem = (kf + kb)/f. As the equilibrium constant determines the equilibrium amount of Red(ads) species, the chemical parameter Kchem represents the rate of removal/resupply of Red(ads) species in the time frame of each potential pulse.
As it has been shown in our recent work [26], for a given value of K and Kchem, voltammetric response is strongly affected by the equilibrium constant (see Fig. 6 in [26] for example). Remarkable voltammetric features emerge for fast electrode reaction. For instance, for Keq > 1, the splitting of the net SW peak is strongly affected by the kinetics of the follow-up chemical step. Shown in Fig. 7 are voltammograms illustrating the effect of Kchem in the case of split net SWV peak. It is important to notice that within a broad region of Kchem values (0.001 < Kchem < 0.5), simultaneous increase of both reduction (forward) and oxidation (backward) current components is encountered. Interestingly, the backward (oxidation) peak current rises more rapidly by increasing Kchem. At the same time, the peak potential of the backward component shifts towards more positive values as Kchem increases. In such scenario, the splitting of the net SW voltammogram vanishes for Kchem of 0.04. In addition, both reduction and oxidation peaks further increase in proportion to Kchem within the interval 0.04 < Kchem < 0.5. Eventually, for Kchem > 0.6, both current components commence decreasing, acquiring typical shape for ECrev mechanism [1,2,3, 26]. Hence, for moderate and high values of Keq, the overall rate of the chemical step influences significantly the electrode reaction. This is also shown in Fig. 8a, where a series of quasi-reversible maxima are presented, simulated for Keq = 10 and several values of Kchem. As the values of the chemical parameter Kchem increase from 0.0075 to 0.075, the position of maximum shifts from log(Kmax) = − 0.775 to log(Kmax) = 1. At the same time, the increasing rate of the follow-up chemical step causes the overall currents to increase, as shown in Fig. 8a. The linear dependence of log(Kchem) vs. log(Kmax) (Fig. 8b) allows determination of the value of Kchem in a very elegant way as described in [26].

Surface ECrev mechanism: case with fast electrode transfer rate: Effect of the dimensionless chemical parameter Kchem to the features of simulated SW voltammograms simulated for a value of the dimensionless electrode kinetic parameter K = 3.0. The equilibrium constant of the follow up chemical reaction was set to Keq = 10, while the value of dimensionless chemical rate parameter Kchem was 0.00001 (a), 0.0075 (b), 0.0255 (c) and 0.5 (d). All other simulation parameters were same as those in Fig. 1

Surface ECrev mechanism. a Quasi-reversible maxima simulated for Keq = 10 and for several values of dimensionless chemical rate parameter Kchem. b Dependence between the logarithm of the chemical rate parameter log(Kchem) and the logarithm of maximal values of the dimensionless electrode kinetic parameters log(Kmax) from Fig. 8a. All other simulation parameters were same as those in Fig. 1
As we have shown in our recent works [25, 26, 37, 39], the observed phenomena in Figs. 7 and 8 arise mainly from the specific chronoamperometric features of this electrode mechanism in SWV. If the rate of electron transfer is comparable to the overall rate of the chemical reaction, simultaneous increase of both forward and backward current components is observed. Similar effect was initially observed in the case of adsorption-complicated ECirev mechanism [42, 43, 48]. Pivotal role in such scenario is attributed to the rate of chemical removal of redox active form Red(ads), causing the electrode reaction to proceed further and the end of potential pulse, thus producing a larger current. The features described in Figs. 7 and 8 are encountered with the surface ECrev mechanism only, and they can be used as diagnostic criteria for qualitative and kinetic characterization as elaborated in detail in [26].
Surface confined electrode reaction coupled with a preceding reversible chemical reaction (surface CrevE)
From all surface redox mechanisms complicated with chemical reactions, the electrode reaction coupled with a preceding chemical step (CrevE mechanism) is, indeed, the most complex one. As the initially available material at the working electrode surface depends on the value of the equilibrium constant Keq, the resupply/removal of Ox(ads) species during the time frame of the experiment is dictated by the chemical rate parameter Kchem. An interplay between the specific chronoamperometric characteristics of the surface electrode reaction and the chemical step results in a complex voltammetric outcome of the experiment [19]. Here, as in the previous case, we focus mainly to voltammetry typical for fast electrode reaction.
Shown in Fig. 9 are theoretical voltammograms of a quasi-reversible electrode reaction, simulated for Keq = 1 and three different values of the chemical rate parameter Kchem. Obviously, an increase of Kchem from 0.01 to 3.16 leads to a simultaneous increase of the overall voltammetric response. Indeed, the effect of the overall chemical kinetics depends strongly on the value of Keq, as shown in Fig. 10, and the overall effect is very complex. The rate of the preceding chemical reaction, represented by the parameter Kchem, influences the intensity, position and the shape of all current components.

Surface CrevE mechanism: case with moderate electron transfer rate: Effect of the dimensionless chemical parameter Kchem to the features of the SW voltammograms simulated for a value of the dimensionless electrode kinetic parameter K = 1.0, and for equilibrium constant of preceding chemical reaction Keq = 1. The values of the dimensionless chemical rate parameter Kchem were 0.01 (a), 0.316 (b) and 3.16 (c). All other simulation parameters are same as those in Fig. 1

Surface CrevE mechanism: case with moderate electron transfer rate: Theoretical SW voltammograms simulated for a value of the dimensionless electrode kinetic parameter K = 0.794, Kchem of 1.0, and for several equilibrium constants of preceding chemical reaction. The values of equilibrium constants Keq are given in the charts
The effect of Kchem is more pronounced when the electrode reaction is fast. Shown in Figs. 11 and 12 are SW voltammograms when the net peak splitting occurs, calculated for Keq = 1 and Keq = 0.01, respectively. Although, at a first glance the situation might look similar, the voltammetric behaviour presented in Figs. 11 and 12 is significantly different. For Keq = 1, the overall rate of the preceding chemical reaction (Kchem ≤ 0.05) has mainly an influence to the peak current of the forward (reduction) component (Fig. 11a–c). However, the rate of the preceding reaction has a remarkable effect to the morphology of both forward and backward current components, in particular at more negative potentials than the peak potentials (see Fig. 11b, c).

Surface CrevE mechanism: case with fast electron transfer rate: Effect of the dimensionless chemical parameter Kchem to the features of the SW voltammograms simulated for a value of the dimensionless electrode kinetic parameter K = 10, and for an equilibrium constant of preceding chemical reaction Keq = 1. The values of the dimensionless chemical rate parameter Kchem were 0.00001 (a), 0.00316 (b), 0.0316 (c), 3.16 (d), 31.6 (e) and 316 (f). All other simulation parameters are same as in Fig. 1

Surface CrevE mechanism: case with fast electron transfer rate: Effect of the dimensionless chemical parameter Kchem to the features of the SW voltammograms simulated for a value of the dimensionless electrode kinetic parameter K = 1.778, and for an equilibrium constant of preceding chemical reaction Keq = 0.01. The values of the dimensionless chemical rate parameter Kchem were 0.001 (a), 0.056 (b), 0.316 (c), 5.62 (d), 56.2 (e) and 562 (f). All other simulation parameters are same as in Fig. 1
Specifically, the current in post-peak potential region of both current components is significantly elevated, the phenomenon being proportional to the rate of the preceding reaction. Such phenomena can be ascribed to the effect of additional resupply of Ox(ads) species by the preceding chemical reaction during the time frame of the voltammetric experiment.
The net SWV peak splitting vanishes in the range 0.05 < Kchem < 10 (Fig. 11c, d). Further increase of Kchem > 10 leads to simultaneous increase of both reduction and oxidation peak currents, while the splitting of net SWV peaks is re-established (see Fig. 11e, f). The voltammetric behaviour of a fast surface electrode reaction preceded by a chemical reaction described in Fig. 11 is unique for this mechanism and can serve as qualitative criteria for characterization of the mechanism.
Furthermore, when the preceding chemical reaction is thermodynamically unfavourable, attributed with Keq = 0.01 (Fig. 12), an initial increase of Kchem from 0.001 to 0.5 leads to disproportional increase of forward and backward current components. Unexpectedly, the oxidation (backward) peak current gains more in the intensity than the reduction one (Fig. 12b, c). For Kchem > 2, the splitting disappears (Fig. 12d), while further increase of Kchem up to 500 causes both current components to gain in the intensity almost identically. For Keq = 0.01, the net SWV peak splitting is lost and cannot be re-established by further increase of Kchem, contrary to the case observed for higher Keq values (compare Figs. 11e, f and 12e, f, for example). Accordingly, the differences of the voltammetric behaviour illustrated in Figs. 11 and 12 can serve as qualitative criteria for rough assessment of the equilibrium constant.
Although the evolution of the voltammetric pattern presented in Fig. 12 resembles the surface ECrev mechanism illustrated in Fig. 7, yet the two cases differ significantly. While the phenomenon of post-peak elevated current for both forward and backward current components is typical for the surface CrevE mechanism (due to the resupply of an electroactive material), it does not appear in the case of ECrev mechanism (compare Fig. 12b–d with Fig. 7, for example). The surface CrevE mechanism differs from the surface ECrev mechanism in, at least, one more relevant aspect. While for the surface ECrev mechanism, the position of quasi-reversible maximum is a function of Kchem (see Fig. 8a), the position of the maximum does not depend on Kchem in the case of surface CrevE mechanism [19].
Shown in Fig. 13a is the dependence of the peak current ratio of the absolute values of the forward and backward components as a function of log(Kchem), estimated for several values of Keq. The expected sigmoid dependences, common for this mechanism [1,2,3,4, 19], exist only for Keq > 1. However, for Keq < 1, the sigmoid curves on Fig. 13a feature a local minimum, roughly in the regions of − 1 < log(Kchem) < 0.1. The linear portions of sigmoidal curves of Fig. 13a are expanded in Fig. 13b. The slope of the linear part of the function |Ψreduction/Ψoxidation| vs. log(Kchem) is inversely proportional to the value of Keq. The latter can be additionally used as a criterion for recognizing the CrevE mechanism. Finally, as explained in [19], the method of measuring the potential separation of the split net peaks as a function of log(Kchem) can be explored for the determination of the chemical rate parameter Kchem at the CrevE mechanism, providing that the values of dimensionless electrode kinetic parameter K and Keq are known.

Surface CrevE mechanism: case with fast electron transfer rate. a Dependence of the ratios of absolute values of reduction/oxidation SWV peak currents as a function of the logarithm of dimensionless chemical parameter Kchem. The curves are calculated for four different values of the equilibrium constant of preceding chemical reaction Keq (Keq values are given in the chart). b Shows the corresponding dependences of the linear part of sigmoidal curves from panel a. The SW voltammograms were simulated for a value of the dimensionless electrode kinetic parameter K = 0.316. All other simulation parameters are same as in Fig. 1
Conclusions
Electrochemical reactions comprise energy exchange between redox active species and an electrically conductive material (working electrode). The kinetics of all electrochemical reactions depends on the type of electrode material, temperature, concentrations of the reactants involved in the redox processes, but also on the applied potential. Commonly, for constant temperature and defined electrode material, relevant kinetic data are evaluated in time-dependent experiments (i.e. chronoamperometric experiments in which the current is measured at different time or voltammetric experiment in which the scan rate is varied) [1,2,3,4,5,6,7,8,9,10, 17]. As it has been shown in this work, for surface electrode processes complicated with chemical reactions, evaluating relevant kinetic parameters from time-dependent experiments is not an easy task, as the voltammetry is affected by several time-dependent critical parameters. In SWV, the frequency, as the most critical time parameter, affects simultaneously both the kinetics of the electron exchange between the working electrode and the redox couple investigates, but also the kinetics of chemical step, too. Consequently, variation of the frequency gives complex voltammetric patterns that are difficult to be elaborated due to mutual interplay between different kinetic parameters [40, 47,48,49,50,51,52,53,54].
In order to avoid such complexity in real experiments, efforts have been made to develop methods for kinetic measurements with time-independent experiments under conditions of SWV. As shown in the recent work of Mirceski et al. [34], elegant access to the formal rate constant of the electron transfer can be achieved by the method of amplitude-based quasi-reversible maximum, or by measuring the peak potential difference of the forward-to-backward current components [37, 55] under constant scan rate. Accordingly, for all electrode mechanisms presented in this work, specific voltammetric features are encountered enabling kinetic and thermodynamic measurements based on the variation of the chemical agent Y at a constant scan rate and frequency of the voltammetric experiment. Moreover, with the plenty of simulated SW voltammograms presented in this work hints are given for recognizing particular electrode mechanism and qualitative characterization. In the very recent work of Compton et al. [56], the CEC mechanism was considered for the first time in cyclic voltammetry, which will be our next task to be elaborated under condition of SWV.
References
Mirceski V, Komorsky-Lovric S, Lovric M (2007) In: Scholz F (ed) Square-wave voltammetry: theory and application, 2nd edn. Springer, Berlin
Compton RG, Banks CE (2011) Understanding voltammetry, 2nd edn. Imperial College Press, London
Bard AJ, Faulkner LR (2004) Electrochemical methods. Fundamentals and applications, 3rd edition. John Wiley & Sons, Inc, New York
Molina A, Gonzales J (2016) Pulse voltammetry in physical electrochemistry and electroanalysis. In: Scholz F (ed) Monographs in electrochemistry. Springer, Berlin
Osteryoung JG, O’Dea JJ (1986) Square-wave voltammetry, electroanalytical chemistry: a series of advances. Marcel Dekker, Inc, New York
Mirceski V, Gulaboski R (2014) Recent achievements in square-wave voltammetry (a review). Maced J Chem Chem Eng 33:1–12
Mirceski V, Gulaboski R, Lovric M, Bogeski I, Kappl R, Hoth M (2013) Square-wave voltammetry-a review on the recent progress. Electroanal 25:2411–2422
Mirceski V, Lovric M (1997) Split square-wave voltammograms of surface redox reactions. Electroanal 9:1283–1287
O’Dea JJ, Osteryoung J, Osteryoung RA (1981) Theory of square-wave voltammetry for kinetic systems. Anal Chem 53:695–701
O’Dea JJ, Osteryoung J (1993) Characterization of quasi-reversible surface processes by square-wave voltammetry. Anal Chem 65:3090–3097
Arsmstrong FA (2015) Electrifying metalloenzymes. In: Cho AE, Goddar WA III (eds) Metalloproteins: theory, calculations and experiments. CRC Press, Taylor&Francis Group, London
Armstrong FA (2002) Voltammetry of proteins. In: Bard AJ, Stratmann M, Wilson GS (eds) Encyclopedia of electrochemistry. vol. 9. Wiley VCH, Weinheim
Armstrong FA (1997) Applications of voltammetric methods for probing the chemistry of redox proteins. In: Lenaz G, Milazz G (eds) Bioelectrochemistry: principles and practice vol. 5. Birkhauser Verlag AG, Basel
Armstrong FA, Heering HA, Hirst J (1997) Reactions of complex metalloproteins studied by protein-film voltammetry. Chem Soc Rev 26:169–179
Leger C, Bertrand P (2008) Direct electrochemistry of redox enzymes as a tool for mechanistic studies. Chem Rev 108:2379–2438
Barlett PN (2008) Bioelectrochemistry: fundamentals, experimental techniques and application. Wiley, Chichester
Gulaboski R, Mirceski V, Bogeski I, Hoth M (2012) Protein-film voltammetry: electrochemical enzymatic spectroscopy. A review on recent progress. J Solid State Electrochem 16:2315–2328
Mirceski V, Gulaboski R (2001) Surface catalytic mechanism in square-wave voltammetry. Electroanal 13:1326–1334
Gulaboski R, Mirčeski V, Lovrić M, Bogeski I (2005) Theoretical study of a surface electrode reaction preceded by a homogeneous chemical reaction under conditions of square-wave voltammetry. Electrochem Commun 7:515–522
Mirceski V, Gulaboski R (2003) The surface catalytic mechanism: a comparative study with square-wave and cyclic staircase voltammetry. J Solid State Electrochem 7:157–165
Mirceski V, Lovric M, Gulaboski R (2001) Theoretical and experimental study of the surface redox reaction involving interactions between the adsorbed particles under conditions of square-wave voltammetry. J Electroanal Chem 515:91–100
Gulaboski R, Mirceski V (2015) New aspects of the electrochemical-catalytic (EC’) mechanism in square-wave voltammetry. Electrochim Acta 167:219–225
Gulaboski R, Mihajlov L (2011) Catalytic mechanism in successive two-step protein-film voltammetry-theoretical study in square-wave voltammetry. Biophys Chem 155:1–9
Gulaboski R (2009) Surface ECE mechanism in protein-film voltammetry-a theoretical study under conditions of square-wave voltammetry. J Solid State Electrochem 13:1015–1024 SupplementaryMaterial: https://springerlink.bibliotecabuap.elogim.com/article/10.1007/s10008-008-0665-5
Gulaboski R (2019) Theoretical contribution towards understanding specific behaviour of “simple” protein-film reactionsinsquare-wavevoltammetry. Electroanal 31:545–553
Gulaboski R, Janeva M, Maksimova V (2019) New aspects of protein-filmvoltammetry of redox enzymes coupled to follow-up reversible chemical reaction in square-wave voltammetry. Electroanal 31. https://doi.org/10.1002/elan.201900028
Gulaboski R, Lovric M, Mirceski V, Bogeski I, Hoth M (2008) Protein-film voltammetry: a theoretical study of the temperature effect using square-wave voltammetry. Biophys Chem 137:49–55
Gulaboski R, Lovric M, Mirceski V, Bogeski I, Hoth M (2008) A new rapid and simple method to determine the kinetics of electrode reactions of biologically relevant compounds from the half-peak width of the square-wave voltammograms. Biophys Chem 138:130–137
Mirceski V, Gulaboski R, Kuzmanovski I (1999) MATHCAD-a tool for numerical calculation of square-wave voltammograms. Bull Chem Technol Maced 18:57–64
Komorsky Lovric S, Lovric M (1995) Kinetic measurements of a surface confined redox reaction. Anal Chim Acta 305:248–255
Song P, Fisher AC, Wadhawan JD, Cooper JJ, Ward HJ, Lawrence NS (2016) A mechanistic study of the EC’ mechanism-the split wave in cyclic voltammetry and square-wave voltammetry. RSC Adv 6:70237–70242
Bonazzola C, Gordillo (2016) Advanced analysis for electrode kinetics studies of surface reactions by applying square-wave voltammetry. Electrochim Acta 213:613–619
Laborda E, Gonzales Molina A (2014) Recent advances on the theory of pulse techniques: a mini review. Electrochim Acta 43:25–30
Mirceski V, Laborda E, Guziejewski D, Compton R (2013) New approach to electrode kinetic measurements in square-wave voltammetry: amplitude-based quasireversible maximum. Anal Chem 85:5586–5594
Dauphin-Durcharme P, Arroyo-Curras N, Kurnik M, Ortega G, Li H, Plaxco KW (2017) Simulation-based approach to determining electron transfer rates using square-wave voltammetry. Langmuir 33:4407–4413
Batchelor-McAuley C, Katelhon E, Barnes EO, Compton RG, Laborda E, Molina A (2015) Recent advances in voltammetry. Chem Open 4:224–260
Mirceski V, Guzijewski D, Lisichkov K (2013) Electrode kinetics measurements with square-wave voltammetry at a constant scan rate. Electrochim Acta 114:667–673
Mirceski V, Guzijewski D, Bozem M, Bogeski I (2016) Characterizing electrode reactions by multisampling the current insquare-wavevoltammetry. Electrochim Acta 213:520–528
Jadresko D, Guziejewski D, Mirceski V (2018) Electrochemical faradaic spectroscopy. ChemElectroChem 5:187–194
Compton RG, Banks CE (2007) Cyclic voltammetry: coupled homogeneous kinetics and adsorption in: understanding voltammetry. Imperial College Press, London
Gulaboski R, Kokoskarova P, Petkovska S (2018) Time-independent methodology to access Michaelis-Menten constant by exploring electrochemical-catalytic mechanism in protein-film cyclic staircase voltammetry. Croat Chem Acta 91:377–382
Mirceski V, Lovric M (2000) Adsorption effects in square-wave voltammetry of an EC mechanism. Croat Chem Acta 73:305–329
Mirceski V, Lovric M (2004) EC mechanism of an adsorbed redox couple. Volume vs surface redox reaction. J Electroanal Chem 565:191–202
Garay F, Lovrić M (2002) Square-wave voltammetry of quasi-reversible electrode processes with coupled homogeneous chemical reactions. J Electroanal Chem 518:91–102
Helfrick JC, Mann MA, Bottomley LA (2016) Diagnostic criteria for the characterization of electrode reactions with chemical reactions following electron transfer by cyclic square wave voltammetry. Electrochim Acta 205:20–28
Garay F, Lovrić M (2002) Quasi-reversible EC reactions at spherical microelectrodes analysed by square-wave voltammetry. J Electroanal Chem 527:85–92
Miler AB, Compton RG (2000) Simulation of square-wave voltammetry. EC and ECE electrode processes. J Phys Chem B 104:5331–5342
Mirceski V (2001) Square-wave voltammetry of an EC reaction of a partly adsorbed redox couple. J Electroanal Chem 508:138–149
Jadresko D, Zelić M (2014) Cyclic multipulse voltammetric techniques. Part II: EC mechanism. J Electroanal Chem 714-715:30–37
Cizmek L, Komorsky-Lovric S, Lovric M (2015) Comparison of cyclic and square-wave voltammetry of irreversible EC mechanism. ChemElectroChem 2:2027–2031
Vettorelo SN, Garay F (2018) Theory of square-wave catalytic adsorptive stripping voltammetry. How to obtain mechanistic information from experimental data. J Electroanal Chem 826:125–132
Vettorelo SN, Garay F (2016) Adsorptive square-wave voltammetry of quasi-reversible electrode processes with a coupled catalytic chemical reaction. J Solid State Electrochem 20:3271–3278
Komorsky-Lovric S, Lovric M (1995) Square-wave voltammetry of quasireversible surface redox reactions. J Electroanal Chem 384:115–122
Lovrić M, Jadresko D (2010) Theory of square-wave voltammetry of quasireversible electrode reactions using an inverse scan direction. Electrochim Acta 55:948–951
Guziejewski D, Mirceski V, Jadresko D (2015) Measuring the electrode kinetics of surface confined electrode reactions at a constant scan rate. Electroanal 27:67–73
Lopez-Tenez M, Laborda E, Molina A, Compton RG (2019) Guidelines for the voltammetric study of electrode reactions with coupled chemical kinetics at an arbitrary electrode geometry. Anal Chem 91:6072–6079
Acknowledgments
We also thank Msci Milkica Janeva for her contribution.
Funding
Valentin Mirceski received support through the NATO Grant No. SPS G5550. Rubin Gulaboski thanks Goce Delcev University Stip for the support via University founded project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gulaboski, R., Mirceski, V. & Lovric, M. Square-wave protein-film voltammetry: new insights in the enzymatic electrode processes coupled with chemical reactions. J Solid State Electrochem 23, 2493–2506 (2019). https://doi.org/10.1007/s10008-019-04320-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-019-04320-7