
Abstract
A glassy carbon electrode (GCE) modified with carbon Printex 6L (Printex6L/GCE) as a novel sensor is proposed. A morphological study was carried out using scanning electron microscopy, and an electrochemical characterization of the proposed electrode was performed by cyclic voltammetry (CV) using [Fe(CN)6]4− as a redox probe. With the incorporation of the carbon Printex 6L film onto the GCE surface, the [Fe(CN)6]4− analytical signal was substantially increased and the difference between the oxidation and reduction potentials (ΔE p) decreased, a characteristic of the electrocatalytic effect. Furthermore, the use of carbon Printex 6L film resulted in an 84 % increase in the oxidation current and a 123 % increase in the reduction current. Faster charge transfer was observed at the proposed electrode/electrolyte interface during CV when compared with GCE. The Printex6L/GCE was tested for ranitidine (RNT) sensing and showed a decrease in the working potential and an increase in the analytical signal, when compared with GCE, again demonstrating an electrocatalytic effect. Under optimized experimental conditions, the developed square-wave adsorptive anodic stripping voltammetry (SWAdASV) method presented an analytical curve that was linear in RNT concentration range from 1.98 × 10−6 to 2.88 × 10−5 mol L−1 with a detection limit of 2.44 × 10−7 mol L−1. The developed Printex6L/GCE was successfully applied to the determination of RNT concentrations in human body fluid samples (urine and serum).
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Several carbon-based materials, including graphite [1, 2], carbon nanofibers [3, 4], carbon nanotubes [5–7], fullerenes [8, 9], graphene [10], diamond-doped boron [11, 12], nanodiamonds [13, 14], and carbon black (CB) [15–22], have been studied for use in the development of electrochemical sensors. Among these carbon materials, CB is often incorrectly called soot. In 2001, Watson and Valberg [23] reported the physical and chemical differences between these compounds and demonstrated that soot has a heterogeneous composition, containing large fractions of organic carbon, while CB consists of small fine particles composed mainly of elemental carbon with low amounts of organic and inorganic surface compounds [23–27]. CB is produced by partial combustion or thermal decomposition of gaseous or liquid hydrocarbons under controlled conditions. Changes in the synthesis conditions can produce a variety of CB with different properties, such as surface area, conductivity, size, and particle structure [23–27].
CB material is widely used in research involving fuel cells [28–30], lithium-ion batteries [31–33], and oxygen reduction reactions [34–38]. Positive, reliable results have been reported in some studies employing CB as an electrochemical sensor [39–41]. Recently, Vicentini et al. [15] studied the behavior of CB (Vulcan-X) and observed a significant improvement in the analytical signal for determining acetaminophen and dopamine concentrations when compared with GCE and edge-plane pyrolytic graphite electrode.
A screen-printed electrode (SPE) modified with CB and Au nanoparticle (AuNP) composite was studied by Arduini et al. [42]. In this work, the authors presented, for the first time, the electrocatalytic effectiveness of AuNPs–CB modified SPE (AuNP–CB–SPE) in electrochemical processes involving different electroactive species, namely, glucose, hydrogen peroxide, hydroquinone (HQ), and ascorbic acid (AA).
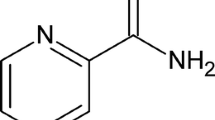
The synthetic compound ranitidine (RNT) (Fig. 1) plays a significant role in the secretion inhibition of histamine H2 receptors from parietal cells. This compound is widely used for the treatment of ulcers in stomach and duodenum in addition to treating other conditions such as indigestion and heartburn [43, 44].

Molecular structure of ranitidine
Several analytical methods for determining RNT in pharmaceutical formulations and biological fluids, such as high-performance liquid chromatography with UV detection [45, 46], near-infrared spectroscopy [47, 48], spectrophotometry UV–Vis [49, 50], and electroanalytical methods [51–58], have been reported. Some of these techniques make use of large volumes of organic solvents that are often toxic. Other techniques involve many steps prior to the final analysis or the development of modified electrodes which require complicated and time-consuming preparation. Moreover, the use of mercury electrodes has been replaced due to environmental concerns.
Thus, in the present work, we have developed a GCE modified with CB (carbon Printex 6L) as a new sensor architecture (Printex6L/GCE) to be used for the electrochemical determination of RNT in biological samples using square-wave adsorptive anodic stripping voltammetry (SWAdASV).
Experimental
Reagents and solutions
Carbon Printex 6L was purchased from Evonik Degussa Brazil Ltda. The stock solution of 1.00 × 10−2 mol L−1 RNT (Sigma-Aldrich) was prepared daily in water, and for dilutions, the chosen supporting electrolyte was used. All reagents used were of analytical grade. The solutions were prepared using ultrapure water of resistivity not less than 18 MΩ cm obtained with a Millipore Milli-Q system.
Synthetic urine samples were prepared according to Laube et al. and containing most of the potential interferents that are present in real samples [59]. The components used for preparation of synthetic urine were NaCl, KCl, CaCl2·2H2O, Na2SO4, KH2PO4, NH4Cl, urea, and water. This solution was used immediately after its preparation.
The synthetic human serum sample was prepared as described by Parham and Zargar [60] with the following composition: NaCl, NaHCO3, glycine, tryptophan, tyrosine, serine, phenylalanine, lysine, histidine, aspartic acid, arginine, alanine, and water. This solution also was used immediately after its preparation. The synthetic samples were spiked with two different concentrations of RNT, and 200 μL aliquots of each sample were added to the electrochemical cell containing the supporting electrolyte for the measurement.
Preparation of the Printex6L/GCE
The GCE was carefully polished with alumina on a polishing cloth and rinsed thoroughly with ultrapure water. It was then sonicated in isopropyl alcohol followed by ultrapure water, for 5 min each, and then dried at room temperature (25 ± 1 °C).
The carbon Printex 6L suspension was prepared by adding 1.0 mg of carbon Printex 6L to 1.0 mL of water, and the mixture was sonicated for 30 min to give a black dispersion. Next, 8 μL of that dispersion was dropped onto the surface of the clean GCE and the solvent was evaporated at 25 °C for 2 h to give the proposed modified GCE, Printex6L/GCE.
Apparatus
All electrochemical measurements were carried out using a μAutolab potentiostat/galvanostat (EcoChemie BV) controlled with the GPES 4.9 software. For the measurements, a three-electrode cell system (volume of 10 mL) was used, with a modified GCE as the working electrode (∅ = 3 mm), a platinum plate as the counter electrode, and an Ag/AgCl (3.0 mol L−1 KCl) electrode as the reference electrode, to which all electrode potentials hereinafter are referred. The square-wave voltammograms were baseline-corrected by the moving average method and smoothed with a Savicky and Golay algorithm using the GPES 4.9 software.
The film morphology was characterized by scanning electron microscopy (SEM) with a Supra 35-VP microscope (Carl Zeiss, Germany) and electron beam energy of 25 keV.
Analytical procedure
The Printex6L/GCE was electrochemically characterized using the [Fe(CN)6]4− probe in 0.1 mol L−1 KCl and cyclic voltammetry (CV).
Impedance spectroscopy (EIS) measurements were carried out at the formal potential of the [Fe(CN)6]4− redox probe, from 10 mHz to 100 kHz (10 points per decade) and with a 10 mV (r.m.s.) ac perturbation, for a 1.0 mmol L−1 [Fe(CN)6]4− in 0.1 mol L−1 KCl solution.
The parameters that influence the SWAdASV techniques, such as pre-concentration potential and time, were evaluated and optimized. Analytical curves were constructed by the addition of different concentrations of the RNT standard solutions. The detection limit was calculated as three times the standard deviation for the blank solution (n = 10) divided by the slope of the analytical curve. The RNT concentration of each biological sample was determined in triplicate by interpolation in the analytical curve previously obtained.
Results and discussion
Scanning electron microscopy
Figure 2 presents a SEM image of carbon Printex 6L on the GCE surface at a magnification of 200,000×. This figure demonstrates that the carbonaceous material is present as beads with dimensions from 30 to 65 nm. Moreover, the carbon Printex 6L formed a homogeneous film and was uniformly distributed on the GCE surface.

SEM image of carbon Printex 6L film on the GCE surface at 200,000×
Characterization of Printex6L/GCE
Initially, the GCE and Printex6L/GCE were characterized electrochemically using the redox [Fe(CN)6]4− as a probe. Figure 3 presents the cyclic voltammograms for a 1.0 × 10−4 mol L−1 [Fe(CN)6]4− solution in 0.1 mol L−1 KCl employing the GCE and Printex6L/GCE. As can be seen, a well-defined pair of redox peaks with a peak separation (ΔE p) of approximately 252 and 56 mV (vs. Ag/AgCl (3.0 mol L−1 KCl)) was obtained for GCE and Printex6L/GCE, respectively. The probe showed a reversible behavior [61] when the Printex6L/GCE was used, which indicates that the proposed electrode promotes a rapid electron transfer. Moreover, the Printex6L/GCE (Fig. 3, red line) provided higher anodic and cathodic current peaks (I pa = 5.97 μA and I pc = −7.08 μA) than the GCE (Fig. 3, black line, I pa = 3.24 μA and I pc = −3.17 μA).

Cyclic voltammograms for 1.0 × 10−4 mol L−1 [Fe(CN)6]4− in 0.1 mol L−1 KCl solution using the GCE (black line) and Printex6L/GCE (red line). v = 50 mV s−1
The carbon Printex 6L formed a highly conductive film on the GCE surface and the proposed film provided an increase in the electrode electroactive area, as presented below. Applying the Randles–Sevcik equation (Eq. 1) [61], the electroactive areas of GCE and Printex6L/GCE were estimated using cyclic voltammetry, which varied the scan rate potential from 10 to 500 mV s−1 (see Fig. S1a, b, in the supplementary data), for a 4.76 × 10−4 mol L−1 [Fe(CN)6]4− in 0.1 mol L−1 KCl solution:
where I p is the anodic or cathodic peak current (A), n is the number of electrons involved in the redox process, A is the electroactive surface area (cm2), D is the diffusion coefficient of [Fe(CN)6]4− (7.60 × 10−6 cm2 s−1) [61], C is the concentration of [Fe(CN)6]4− in bulk solution (4.76 × 10−7 mol cm−3), and v is the potential scan rate (V s−1). The slopes of I p versus v 1/2 for the anodic process (see Fig. S1a, b insets, in the supplementary data) were 1.69 × 10−5 and 2.77 × 10−5 A s1/2 V−1/2 for GCE and Printex6L/GCE, respectively. The calculated electroactive areas were 0.048 cm2 for GCE and 0.079 cm2 for Printex6L/GCE; thus, the carbon Printex6L increased the area by a factor of 1.6 over GCE.
Furthermore, to evaluate the ability of the proposed electrode to transfer electrons, the heterogeneous electron transfer rate constant (k 0) was calculated for the GCE and the Printex6L/GCE using the Nicholson equation [62] for quasi-reversible systems controlled by diffusion:
where ψ is a kinetic parameter, k 0 is the heterogeneous electron transfer rate constant (cm s−1), π is the mathematical constant (3.1415), D is the diffusion coefficient of [Fe(CN)6]4− (7.60 × 10−6 cm2 s−1), n is the number of electrons involved in the redox process, v is the potential scan rate (V s−1), F is the Faraday constant (96,485 C mol−1), R is the Universal gas constant (8.314 J K−1 mol−1), and T is the thermodynamic temperature (298.15 K).
The ψ values were calculated by the Lavagnini equation [63], which relates the potential scan rate with a ΔE p value:
So, the calculated k 0 was 6.55 × 10−4 cm s−1 for GCE and 1.21 × 10−1 cm s−1 for Printex6L/GCE, a 185 times increase for the proposed electrode, indicating a significant improvement in the electron transfer rate on the Printex6L/GCE. This can be attributed to the higher electrical conductivity provided by the carbon Printex 6L.
Additionally, the GCE and Printex6L/GCE were also characterized by EIS using a 1.0 × 10−3 mol L−1 [Fe(CN)6]4− solution prepared in 0.1 mol L−1 KCl. Figure 4 shows the resulting Nyquist plots for GCE and Printex6L/GCE. The catalytic properties of the electrode/electrolyte interface are responsible for the obtained values of charge transfer resistance.

Nyquist plots of (red squares) GCE and (black circles) Printex6L/GCE. The measurements were performed in 0.1 mol L−1 KCl solution containing 1.0 × 10–3 mol L−1 of the redox couple [Fe(CN)6]4− in a frequency range from 0.01 Hz to 100 kHz
Analyzing the impedance spectra (Fig. 4, red line) for GCE, we observed a well-defined semi-circle at higher frequencies, which decreases substantially for Printex6L/GCE (Fig. 4, black line). This behavior is evidence that the electron transfer resistance decreased dramatically when Printex6L/CGE was used, which can result in greatly improved electron transfer on the electrode surface.
The excellent conductivity of carbon Printex 6L decreases the resistance in the electrode/solution system [64]. The quantity of oxygen atoms in the carbon Printex 6L provides a larger number of active sites on the electrode surface for analyte adsorption which can be related to the decrease in resistance [65]. These results also are in agreement with our calculated k 0 values, using CV, and demonstrated that the carbon Printex 6L film was successfully adhered to the GCE surface.
Electrochemical oxidation of RNT on the Printex6L/GCE
Characterization of the electrochemical behavior of the RNT at the Printex6L/GCE was carried out by CV in 0.2 mol L−1 phosphate buffer (pH 4.5). As demonstrated in Fig. 5, an anodic peak was obtained at +0.98 V for the Printex6L/GCE in the presence of 9.90 × 10−5 mol L−1 RNT solution. No reduction peak was observed, indicating an irreversible oxidation process for the RNT at the electrode surface. Using the same experimental conditions, the electrochemical response for RNT on a GCE was also evaluated (Fig. 5). The GCE presented an anodic peak for RNT oxidation at +1.14 V.

Cyclic voltammograms for a 9.90 × 10−5 mol L−1 RNT in 0.2 mol L−1 phosphate buffer (pH 4.5) solution, using the GCE (black line) and the Printex6L/GCE (red line). v = 50 mV s−1
Comparing the results, it is possible to observe an increase in the analytical signal from 1.5 μA for GCE to 16.5 μA when the Printex6L/GCE was used. Moreover, there was a negative shift of 160 mV in the anodic peak potential when the Printex6L/GCE was employed, indicating an electrocatalytic effect of carbon Printex 6L on the RNT oxidation, which demonstrates a great ability for RNT detection.
Effect of scan rate
The effect of the scan rate on the electrochemical response of the RNT at the Printex6L/GCE was carried out by cyclic voltammetry in 0.2 mol L−1 phosphate buffer (pH 4.5). For this, CV assays were carried out at different scan rate potentials (from 10 to 500 mV s−1) in the presence of 9.90 × 10−5 mol L−1 RNT to evaluate RNT oxidation.
There was a shift in the anodic peak potentials with increasing scan rate potentials (data not shown). A linear relationship (r = 0.989) was obtained between the logarithm of the oxidation peak current (log I p) and the logarithm of the scan rate (log v), with a slope of 0.76. According to previous studies [66, 67], this value indicates that the RNT oxidation process on the Printex6L/GCE surface can be controlled for both process, diffusion or adsorption of the species, i.e., there is a mixing process, since the obtained value is between 0.5 (diffusion-controlled process) and 1.0 (adsorption-controlled process).
In order to better understand the RNT oxidation onto Printex6L/GCE surface, plots were constructed between the anodic peak current (I ap) and the scan rate potential (v) and between the anodic peak current (I ap) and the square root of the scan rate (v 1/2). According the cyclic voltammetry theory, a linear relationship between I ap versus v suggests an adsorption-controlled process. On the other hand, a linear relationship between I ap versus v 1/2 characterizes a diffusion-controlled process [66, 67]. The RNT oxidation process using the proposed electrode presented a linear relationship by the dependence of I ap versus v, according to the regression equation (Eq. 4) showing the prevalence of an adsorption-controlled process:
This behavior was also observed by Xi and Ming [58] in a recent publication.
Effect of the supporting electrolyte and pH
The effects of the pH and the composition of the supporting electrolyte on the electrochemical determination of RNT using the Printex6L/GCE were investigated. Initially, the effect of pH was evaluated in the range from 3.5 to 8.0 in 0.2 mol L−1 phosphate buffer solution by SWAdASV, for a 2.97 × 10−5 mol L−1 RNT solution. The highest analytical signal and better peak definition were obtained at pH 4.5, so this pH was selected for further experiments.
The composition of the supporting electrolyte was also evaluated using phosphate, acetate, and Britton–Robinson buffer solutions all fixed at pH 4.5. The highest analytical signal and better shape for the anodic peak current were observed for the phosphate buffer solution, which was employed for further experiments.
Effect of pre-concentration potential and time
The study of pre-concentration potential on the oxidation peak current of the Printex6L/GCE was assessed for a 2.97 × 10−5 mol L−1 RNT solution in 0.2 mol L−1 phosphate buffer (pH 4.5), in the range from −0.5 to 0.9 V with a pre-concentration time of 120 s. The anodic peak current increased up to 0.3 V when the pre-concentration potential was varied from −0.5 to 0.3 V (see Fig. S2a in the supplementary data). However, this pre-concentration potential was not reproducible, so a pre-concentration potential of 0.5 V, which presented better repeatability and a good voltammetric profile, was utilized for further studies.
Beyond that, the effect of pre-concentration time was also evaluated in the range from 20 to 180 s. The anodic peak current increased up to 120 s, and for longer times remained practically constant (see Fig. S2b in the supplementary data). After 2 min, there was a maximum of saturation by the RNT molecules on the Printex6L/GCE surface. Thus, for the RNT pre-concentration step, we selected a pre-concentration potential of 0.5 V and a pre-concentration time of 120 s.
Optimization of parameters and analytical curve
The SWAdASV parameters, square-wave frequency (f), pulse amplitude (a), and scan increment (ΔE S) were optimized for a 9.90 × 10−6 mol L−1 RNT solution in 0.2 mol L−1 phosphate buffer (pH 4.5). The studied ranges were 10 Hz ≤ f ≤ 70 Hz, 10 mV ≤ a ≤ 180 mV, and 1.0 mV ≤ ΔE S ≤ 10 mV. The values were selected (f = 30 Hz, a = 120 mV, and ΔE S = 5.0 mV), taking into account repeatability, baseline stability, accuracy, and magnitude of analytical signal onto the Printex6L/GCE for RNT determination.
After optimization of the SWAdASV parameters, the analytical curve for RNT using the Printex6L/GCE was carried out by adding, in the electrochemical cell, successive aliquots of RNT concentrations ranging from 1.98 × 10−6 to 2.88 × 10−5 mol L−1, and the obtained SW voltammograms are shown in Fig. 6. The analytical curve (Fig. 6, inset) showed a linear concentration range with the following linear regression equation: I p (μA) = 5.9 ± (0.9) × 10−8 + 0.84 ± (0.01) [RNT] (× 10–6mol L−1); r = 0.993 with a detection limit of 2.44 × 10−7 mol L−1 (calculated as described in experimental section).

SW voltammetric curves at a Printex6L/GCE for different concentrations of RNT in 0.2 mol L−1 phosphate buffer (pH 4.5): (1) blank, (2) 1.98, (3) 3.94, (4) 5.91, (5) 7.86, (6) 11.7, (7) 19.4, and (8) 28.8 × 10– 6 mol L−1. SWAdASV parameters: f = 30 Hz, a = 120 mV, ∆E s = 5 mV. Inset: Corresponding analytical curve
Table 1 presents the analytical parameters of the proposed method compared with other electroanalytical procedures reported in the literature. The proposed Printex6L/GCE presented a detection limit lower than those reported for some other RNT sensors [54, 55, 57], while it was similar to the sensor proposed by Ming and Xi [58] yet higher than that reported by some others [52, 53, 56]. However, the sensors developed by Norouzi et al. [53] and Rezaei et al. [56] employed modified electrodes and are complicated and time-consuming to prepare. Moreover, the procedure reported by Malagutti and Mazo [52] used a mercury electrode, which has since been replaced because of environmental issues. In addition, the here-proposed sensor does not require a prior modification step and is easy to prepare.
The intra-day repeatability (n = 10) of one Printex6L/GCE was investigated for two different levels of RNT solution (3.98 × 10−6 and 1.19 × 10−5 mol L−1) in 0.2 mol L−1 phosphate buffer (pH 4.5). Relative standard deviations (RSDs) of 4.27 and 4.33 % were obtained, respectively, indicating a good stability of the proposed film. In addition, the repeatability of the preparation of three different Printex6L/GCE was evaluated from the SWAdASV measurements for a 3.98 × 10−6 mol L−1 RNT in 0.2 mol L−1 phosphate buffer solution (pH 4.5). A RSD of 3.55 % was obtained, showing a high repeatability of the electrode fabrication that is related to the ease preparation of the Printex6L/GCE.
Determination of RNT in human body fluid samples
Finally, the determination of RNT in human body fluid samples (urine and serum) was performed using the proposed method and the Printex6L/GCE. Table 2 presents the results for the analysis of recovery percentage in the samples. The Printex6L/GCE provided an accurate response for RNT, even in the presence of the complex matrices, with recoveries that ranged from 91.7 to 106 %. Furthermore, electrode fouling/response attenuation was not detected in the complex matrices, demonstrating the potential application of this method for the determination of RNT in biological samples.
Conclusions
In this study, the carbon Printex 6L was immobilized in GCE and this new proposed electrode was applied for RNT determination in biological samples using a SWAdASV method. The incorporation of the carbon Printex 6L film on the GCE surface produced an increase in the analytical signal for the redox [Fe(CN)6]4− probe and a decrease in the oxidation and reduction potentials when compared with GCE. Thereafter, faster charge transfer was observed at the proposed electrode/electrolyte interface during CV when compared with GCE.
The Printex6L/GCE presented an electrocatalytic effect on RNT oxidation, leading to a reduction in the working potential and increasing the analytical signal when compared to the GCE. The proposed sensor was applied to the determination of RNT by SWAdASV in the range from 1.98 × 10−6 to 2.88 × 10−5 mol L−1 with a detection limit of 2.44 × 10−7 mol L−1.
Intra- and inter-day repeatability studies showed excellent stability of the carbon Printex 6L film. Furthermore, the proposed SWAdASV method was used for RNT determination in spiked human body fluid samples (urine and serum) with satisfactory recoveries, indicating the promising potential applications of Printex6L/GCE in evaluating biological samples.
References
Teixeira MFS, Marcolino-Junior LH, Fatibello-Filho O, Dockal ER, Cavalheiro ETG (2004) J Braz Chem Soc 15(6):803–808
Zhao X, Zhuang Q-C, Shi Y-L, Zhang X-X (2015) J Appl Electrochem 45(9):1013–1023
Ding W, Wu M, Liang M, Ni H, Li Y (2015) Anal Lett 48(10):1551–1569
Thamer BM, El-Newehy MH, Al-Deyab SS, Abdelkareem MA, Kim HY, Barakat NAM (2015) Appl Catal A 498:230–240
Vicentini FC, Silva TA, Pellatieri A, Janegitz BC, Fatibello-Filho O, Faria RC (2014) Microchem J 116:191–196
Oliveira GG, Janegitz BC, Zucolotto V, Fatibello-Filho O (2013) Cent Eur J Chem 11(11):1837–1843
Vicentini FC, Figueiredo-Filho LCS, Janegitz BC, Santiago A, Pereira ER, Fatibello-Filho O (2011) Quim Nova. 34(5):825–830
Barberis A, Spissu Y, Fadda A, Azara E, Bazzu G, Marceddu S, Angioni A, Sanna D, Schirra M, Serra PA (2015) Biosens Bioelectron 67:214–223
Gao YF, Yang T, Yang XL, Zhang YS, Xiao BL, Hong J, Sheibani N, Ghourchian H, Hong T, Moosavi-Movahedi AA (2014) Biosens Bioelectron 60:30–34
Chekin F, Bagheri S, Abd Hamid SB (2014) J Solid State Electrochem 18(4):893–898
Lourencao BC, Medeiros RA, Thomasi SS, Ferreira AG, Rocha-Filho RC, Fatibello-Filho O (2016) Sensor Actuat B-Chem 222:181–189
Lourencao BC, Baccarin M, Medeiros RA, Rocha-Filho RC, Fatibello-Filho O (2013) J Electroanal Chem 707:15–19
Briones M, Casero E, Petit-Dominguez MD, Ruiz MA, Parra-Alfambra AM, Pariente F, Lorenzo E, Vazquez L (2015) Biosens Bioelectron 68:521–528
Liu L, Song C, Zhang Z, Yang J, Zhou L, Zhang X, Xie G (2015) Biosens Bioelectron 70:351–357
Vicentini FC, Ravanini AE, Figueiredo-Filho LCS, Iniesta J, Banks CE, Fatibello-Filho O (2015) Electrochim Acta 157:125–133
Huang Y, Yan H, Tong Y (2015) J Electroanal Chem 743:25–30
Sun J, Gan T, Meng W, Shi Z, Zhang Z, Liu Y (2015) Anal Lett 48(1):100–115
Noskova GN, Zakharova EA, Kolpakova NA, Kabakaev AS (2012) J Solid State Electrochem 16(7):2459–2472
JB Eastwood, AP Christensen, DR Armstrong, RN Bates J Solid State Electrochem 3(4):179–186
Fu Y, Liu Y, Li Y, Li J, Qiao J, Zhang J (2015) J Solid State Electrochem 19(11):3355–3363
Miranda-Hernández M, Ayala J, ME Rincón J Solid State Electrochem 7(5):264–270
Zhutaeva GV, Bogdanovskaya VA, Davydova ES, Kazanskii LP, Tarasevich MR (2013) J Solid State Electrochem 18(5):1319–1334
Watson AY, Valberg PA (2001) Am Ind Hyg Assoc J 62(2):218–228
Wang M-J, Gray CA, Reznek SA, Mahmud K, Kutsovsky Y (2000) Carbon black. In: Kirk-Othmer Encyclopedia of Chemical Technology. John Wiley & Sons, New York
Long CM, Nascarella MA, Valberg PA (2013) Environ Pollut 181:271–286
Bello D, Hsieh S-F, Schmidt D, Rogers E (2009) Nanotoxicology 3(3):249–261
Fitzer E, Kochling KH, Boehm HP, Marsh H (1995) Pure Appl Chem 67(3):473–506
Kaluza L, Larsen MJ, Zdrazil M, Gulkova D, Vit Z, Solcova O, Soukup K, Kostejn M, Bonde JL, Maixnerova L, Odgaard M (2015) Catal Today 256:375–383
Huggins TM, Pietron JJ, Wang H, Ren ZJ, Biffinger JC (2015) Bioresour Technol 195:147–153
Jang H, Ocon JD, Lee S, Lee JK, Lee J (2015) J Power Sources 296:433–439
Lin HB, Huang WZ, Rong HB, Hu JN, Mai SW, Xing LD, Xu MQ, Li XP, Li WS (2015) J Power Sources 287:276–282
Bauer W, Noetzel D, Wenzel V, Nirschl H (2015) J Power Sources 288:359–367
Huang KJ, Zhang JZ, Jia YL, Xing K, Liu YM (2015) J Alloys Compd 641:119–126
Biloul A, Contamin O, Scarbeck G, Savy M, Vandenham D, Riga J, Verbist JJ (1992) J Electroanal Chem 335(1–2):163–186
Antonin VS, Assumpcao MHMT, Silva JCM, Parreira LS, Lanza MRV, Santos MC (2013) Electrochim Acta 109:245–251
Barros WRP, Wei Q, Zhang G, Sun S, Lanza MRV, Tavares AC (2015) Electrochim Acta 162:263–270
Reis RM, Valim RB, Rocha RS, Lima AS, Castro PS, Bertotti M, Lanza MRV (2014) Electrochim Acta 139:1–6
Silva FL, Reis RM, Barros WRP, Rocha RS, Lanza MRV (2014) J Electroanal Chem 722:32–37
Talarico D, Arduini F, Amine A, Moscone D, Palleschi G (2015) Talanta 141:267–272
Deroco PB, Vicentini FC, Fatibello-Filho O (2015) Electroanalysis 27(9):2214–2220
Vicentini FC, Raymundo-Pereira PA, Janegitz BC, Machado SAS, Fatibello-Filho O (2016) Sensor Actuat B-Chem 227:610–618
Arduini F, Zanardi C, Cinti S, Terzi F, Moscone D, Palleschi G, Seeber R (2015) Sensor Actuat B-Chem 212:536–543
Brogden RN, Carmine AA, Heel RC, Speight TM, Avery GS (1982) Drugs 24(4):267–303
Goodman LS, Hardman JG, Limbird LE, Gilman AG (2001) Goodman & Gilman’s the pharmacological basis of therapeutics. McGraw-Hill, New York
Berzas Nevado JJ, Castaneda Penalvo G, Rodriguez Dorado RM, Rodriguez Robledo V (2013) J Chromatogr. B: Anal Technol Biomed Life Sci 921:56–63
Kiszkiel I, Starczewska B, Lesniewska B, Pozniak P (2015) J Pharm Biomed Anal 106:85–91
Rosa SS, Barata PA, Martins JM, Menezes JC (2008) Talanta 75(3):725–733
Stepanova EV, Arzamastsev AP, Titova AV (2009) Pharm Chem J 43(7):425–427
Ahmad AKS, Kawy MA, Nebsen M (1999) Anal Lett 32(7):1403–1419
Sastry CSP, Rao SG, Rao J, Naidu PY (1997) Anal Lett 30(13):2377–2390
Altinoz S, Ozer D, Temizer A, Bayraktar Y (1992) Anal Lett 25(1):111–118
Malagutti AR, Mazo LH (2003) J Braz Chem Soc 14(2):274–280
Norouzi P, Ganjali MR, Daneshgar P (2007) J Pharmacol Toxicol Methods 55(3):289–296
Pfaffen V, Ines Ortiz P (2010) Ind Eng Chem Res 49(9):4026–4030
Pereira PAR, Teixeira MFS, Fatibello-Filho O, Dockal ER, Bonifacio VG, Marcolino-Junior LH (2013) Mater Sc eng C 33(7):4081–4085
Rezaei B, Lotfi-Forushani H, Ensafi AA (2014) Mater Sc eng C 37:113–119
Richter P, Toral MI, Munozvargas F (1994) Analyst 119(6):1371–1374
Xi X, Ming L (2013) Asian J Chem 25(10):5315–5318
Laube N, Mohr B, Hesse A (2001) J Cryst Growth 233(1–2):367–374
Parham H, Zargar B (2001) Talanta 55:255–262
Bard AJ, L.R. F (2001) Electrochemical methods: fundamentals and applications, 2a ed. John Wiley & Sons, New York
Nicholson RS (1965) Anal Chem 37(11):1351–1355
Lavagnini I, Antiochia R, Magno F (2004) Electroanalysis 16(6):505–506
Liu E, Zhang X (2014) Anal Methods 6(21):8604–8612
Assumpção MHMT, De Souza RFB, Rascio DC, Silva JCM, Calegaro ML, Gaubeur I, Paixão TRLC, Hammer P, Lanza MRV, Santos MC (2011) Carbon 49(8):2842–2851
Gosser DK (1993) Cyclic voltammetry: simulation and analysis of reaction mechanisms. Wiley-VCH, New York
Kissinger PT, Heineman WR (1996) Laboratory techniques in electroanalytical Chemistry, 2a ed. Marcel Dekker, New York
Acknowledgments
The authors are grateful to the following Brazilian funding agencies, FAPESP (Proc. 2013/16770-0 and 2014/03019-7), CNPq (561071/2010-1), and CAPES, for financial support and scholarships.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 529kb)
Rights and permissions
About this article

Cite this article
Silva, L.P., Vicentini, F.C., Lourencao, B.C. et al. A new sensor architecture based on carbon Printex 6L to the electrochemical determination of ranitidine. J Solid State Electrochem 20, 2395–2402 (2016). https://doi.org/10.1007/s10008-016-3143-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3143-5