
Abstract
Novel hierarchical porous carbon aerogels (PCAs) derived from amphiphilic carbonaceous material (ACM) have been mass-prepared via a facile solvent exchange induced self-assembly process and subsequent carbonization and KOH activation. The resulting products are stacked up by highly interconnected carbon nanoparticles with a certain amount of micropores and mesopores, which aggregate to build a three-dimensional macroporous architecture. The hierarchical porous structure facilitates fast ion transportation inside the electrode simultaneously preserving efficient ion surface electrochemical reactions. Capacitive and rate performances were evaluated by fabricating symmetric capacitors with both aqueous and organic electrolytes. The PCA-0.5 and PCA-1.0 electrodes exhibit superior specific capacitances of 261.2 and 227.9 F g−1 at a current density of 0.05 A g−1 in 6 M KOH electrolyte, and still remain 145.5 and 175.4 F g−1 as the current density increases to 100 A g−1, respectively. Remarkably, the PCA-0.5 and PCA-1.0 electrodes show stable cycle durability with a slight capacitance loss of 8.2 and 11.2 % after 5000 cycles at 1 A g−1, respectively. Furthermore, in organic electrolyte system, the PCA-1.0 electrode manifests an outstanding capacitance of 155.4 F g−1 at a current density of 0.05 A g−1. The encouraging results demonstrate that PCAs are a sort of promising and competitive supercapacitors electrode materials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rapid development of portable electronic devices and electric vehicles market has greatly promoted the demand for high-power energy resources. In recently years, supercapacitors or electrical double layer capacitors (EDLC) have attracted extensive attention due to their impressive advantages in high-power density, safe and long cycle life, and become an alternative or supplement for rechargeable batteries [1–5].
Porous carbon materials, such as activated carbon [6, 7], mesoporous carbon [8, 9], porous graphitic carbon [10], and carbon aerogels [11, 12], have been used most frequently as supercapacitor electrode materials because of their low cost, good electrical conductivity, high surface area, and excellent chemical and thermal stability [13]. Among various porous carbon, carbon aerogels (CAs) are considered to be a kind of novel and promising electrode materials for supercapacitors because of their high surface area, adjustable pore and particle size, and unique hierarchical porous networks. CAs possess adequate macropores, which ensue fast ion diffusion for high-power density, and provide abundant micropores and small mesopores, leading to high energy density [14–16]. Typically, carbon aerogels are prepared by the sol–gel polycondensation of resorcinol (R) with formaldehyde (F), followed by organic solvent exchange, supercritical drying, and carbonization. However, there are some key factors limiting the large-scale industrial production of CAs, such as relatively expensive raw materials, time-consuming gelating process, and high cost of supercritical drying device. Hereby, we propose a facile and feasible method aiming at overcoming the abovementioned problems, meanwhile making CAs be competitive with other carbon materials used as electrode materials for supercapacitors.
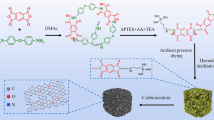
In this paper, amphiphilic carbonaceous material (ACM) is selected as a precursor to prepare carbon aerogels. ACM is a kind of alkaline aqueous solution and polar organic solvent-soluble carbonaceous material, which can be obtained from coal-tar pitch, green needle coke, and petroleum coke by oxidation in liquid phase [17, 18]. As a carbon precursor for carbon aerogels, ACM possesses the preponderances of sufficient surface functional groups, widened d002, and disperses in aqueous solution with nanoscale, which are beneficial to the environment-friendly preparation process. Moreover, the unique microstructure of ACM can shorten the gelation time, reduce the production cost, and make it simple for CAs to be mass produced. Porous carbon aerogels (PCAs) are prepared using ACM as carbon precursor by acetone exchange under ambient pressure followed by carbonization and KOH activation in an inert atmosphere. The morphology and pore structure of PCAs are analyzed by various techniques. To demonstrate the potential of porous carbon aerogels for energy storage, symmetric capacitors of aqueous and organic electrolyte are constructed for evaluating the capacitive and the rate performance.
Experimental
Materials synthesis
Coal-tar pitch (CP) was selected as a raw material to prepare ACM via an oxidation process with a mixture of concentrated nitric acid and sulfuric acid (V/V = 3/7). The details of the procedure were described in our earlier report [18]. ACM of 12 g was added to 40 ml ethylenediamine solution with a pH of 12 and stirred at 80 °C for 1 h. The fully dispersed ACM was then dropwise added into 500 ml acetone and stirred for 1 h. Subsequently, the mixture was sealed and placed under ambient temperature till it separated into two layers. The upper clear liquid was removed, and another 500 ml acetone was refilled into the precipitate and stirred for 10 h to thoroughly remove the residual water. Finally, the precipitate was collected and dried at 50 °C for 12 h to get organic aerogels. After carbonization at 500 °C for 2 h under N2 flow, the obtained carbon aerogels (CAs) were further activated with KOH at 800 °C to get porous carbon aerogels (PCAs). PCAs obtained with the KOH/carbon mass ratio of 0.5 and 1 were named as PCA-0.5 and PCA-1.0, respectively. Samples prepared are named and listed in Table 1.
Characterization
The morphology and microstructure of as-synthesized samples were characterized by FEI NanoSEM 430 scanning electron microscopy (SEM) and Philips Tecnai G2 F20 high-resolution transmission electron microscope (TEM). X-ray photoelectron spectroscopy (XPS) measurement was conducted with a PHI-1600ESCA electron system (America PE Company) using Al Kα (1486.6 eV) radiation over an area of 0.8 mm2 to determine the atomic composition of the sample surface. Raman spectroscopy was carried out using a Thermo Electron Raman microscopy (λ = 532 nm).
The porous structure parameters of the samples were tested by N2 adsorption/desorption at 77 K on Micrometrics ASAP 2020 instrument. The specific surface area (S BET) was obtained using the Brunauer–Emmett–Teller (BET) method, and mesopore surface area (S mes) was calculated by Barrett–Joyner–Halenda (BJH) method from the desorption branch of isotherm. Pore volume was determined from the amount of nitrogen adsorbed at a relative pressure (P/P 0) of 0.99 using the t plot method. Cylindrical density function theory (DFT) method was used to obtain the pore size distribution.
Electrochemical performance
The electrodes for supercapacitors were fabricated by pressing a mixture of carbonaceous materials, polytetrafluoroethylene (PTFE), and acetylene black to the nickel foam current collector with a mass ratio of 80:10:10. To examine the electrochemical performances of the as-prepared electrodes, coin-type cells of symmetrical supercapacitors were assembled using 6 M KOH aqueous solution and 1 M Et4NBF4/PC as electrolytes, respectively. Galvanostatic charge–discharge experiment was performed on Land and Arbin battery test instruments. Cyclic voltammetry (CV) experiments and electrochemical impedance spectroscopy (EIS) were carried out in Princeton PARSTAT2273 electrochemical system. The voltage range of CV was 0–1.0 V for aqueous electrolyte and 0–2.7 V for organic medium, and the Nyquist plots were recorded at the frequency ranged from 100 KHz to 10 mHz.
The specific gravimetric capacitance (C g, F g−1) of the supercapacitors was calculated according to the formula:
where, I presents for the current (A), ∆t for the discharge time (s), ∆V for the voltage (V), and m for the mass (g) of activated carbon in a single electrode.
Results and discussion
Structural characterization
SEM and TEM were utilized to examine the morphology and nanostructure of the as-synthesized samples. As depicted in Fig. 1a, d, the skeletal structure of the CAs consists of interconnected spherical nanoparticles with diameters of 30–50 nm, which aggregate to build a 3D hierarchical mesoporous and macroporous network. Figure 1b, e presents the SEM and TEM images of PCA-0.5 prepared with a KOH/carbon mass ratio of 0.5. In Fig. 1b, PCA-0.5 completely maintains the original 3D porous structure of CAs, and no massive texture is observed in the vision scope due to the low alkali/carbon ratio of activation. TEM image (Fig. 1e) shows the similar result; PCA-0.5 keeps the original shape of CAs and a certain amount of macropores are formed by the interconnected nano-sized carbon monomers. However, as the KOH/carbon mass ratio increased to 1.0, partially fusing and collapsing phenomenon of carbon nanoparticle skeletons happened due to the strong corrosion effect of KOH activation, which is seen in Fig. 1c, f.

SEM and high-resolution TEM images of a, d CAs, b, e PCA-0.5, and c, f PCA-1.0
The surface elemental composition of CAs, PCA-0.5, and PCA-1.0 were estimated by XPS (Table 2). The results show that all the samples are mainly composed by C and a little amount of O, N, and S without any metal impurities. The C atomic percentage of CAs, PCA-0.5, and PCA-1.0 are 85.3, 89.3, and 91.3 %, respectively. PCAs exhibit a relatively high content of C due to the activation process further removing a certain amount of heteroatoms, which may contribute to a better conductivity. The C 1-s spectra are resolved into five peaks (Fig. 2) that correspond to the following carbon bonds: graphitic carbon (C = C, 284.6–285.1 eV), epoxy/hydroxyls (C–O, 286.3–287.0 eV), carbonyl (C = O, 287.5–288.1 eV), carboxyls or ester (O–C = O, 289.3–290.0 eV), and shake-up satellite peaks due to π-π* transitions in aromatic rings (291.2–292.1 eV) [19]. The graphitic carbon of PCA-0.5 and PCA-1.0 are 61.56 and 60.33 %, respectively, which is lower than that of 73.37 % for CAs, indicating PCAs are more disordered than CAs.

XPS C 1-s spectra of samples
Figure 3 shows the Raman spectra of samples. As shown, all the Raman spectra display two bands: the D-band (1300–1500 cm−1) which is related to the structural defects or partially disordered graphitic domains, and the G-band (1550–1620 cm−1) which is associated with graphitic carbons [20]. The intensity ratio of D-band to G-band (I D/I G), which can be used to estimate the relative content of defects of carbon materials, is 0.96, 0.99, and 1.06 for CAs, PCA-0.5, and PCA-1.0, respectively. The result reflects that PCAs possess a higher content of defects than CAs. Besides, PCAs exhibit a relatively narrow full width at half maximum (FWHM) than CAs, indicating a higher graphitization degree and better electrical conductivity of PCAs.

Raman spectrum of samples
Pore structure parameters of CAs, PCA-0.5, and PCA-1.0 were characterized by nitrogen absorption/desorption. Figure 4a shows that the absorption/desorption isotherms of all the three samples are a combination of type II and IV [21] with hysteresis loop and limitless adsorption at high p/p o, which indicate the existence of a certain amount of mesopores and macropores.

a N2 absorption/desorption isotherms of CAs, PCA-0.5, and PCA-1.0 under 77 K. b Pore size distribution of CAs, PCA-0.5, and PCA-1.0
To better understand the pore structures and the pore size distribution of the samples, pores are divided into four types: (A) micropores within carbon nanoparticles with diameters below 2 nm, (B) small mesopores within carbon nanoparticles with diameters from 2 to 5 nm, (C) large mesopores formed from the closely packed carbon nanoparticles with diameters from 5 to 50 nm, and (D) large pores and channels formed from the loosely packed carbon nanoparticles with diameters over 50 nm.
According to the pore size distribution curves (Fig. 4b), all the samples exhibit hierarchical pore structures. The CAs sample possesses channels of type A, C, and D, and little type B pores are observed. Because CAs are piled up by highly interconnected spherical nanoparticles, large mesopores and macropores formed from the closely packed carbon nanoparticles can be detected. Besides, a number of micropores inside carbon nanoparticles were also produced due to the carbonization process. Generally, KOH activation greatly increases the amount of micropores and large mesopores. Compared with CAs, PCA-0.5 not only maintains the pore structures of type C and D but also exhibits a significantly increase of type A channels and the appearance of type B channels. Owing to the relatively low concentration of KOH used for activation, appropriate micropores and small mesopores on the surface of carbon skeleton particles were produced, and the 3D macroporous structures were kept intact at the same time. However, PCA-1.0 displays an increase in type A and especially B channels and a decrease in type C and D channels. As the KOH/carbon mass ratio of activation increased to 1.0, excessive activation agent produced a large number of micropores and small mesopores at the sacrifice of original large mesopores and macropores of CAs.
The porous structure parameters of the synthesized samples are summarized in Table 3. Among them, the CAs sample only has a specific surface area of 366.1 m2 g−1. As the mass ratio of KOH/carbon increases from 0.5 to 1, the S BET of PCAs increase from 1246.0 to 2435.0 m2 g−1.
Electrochemical performance
Cyclic voltammetry measurement was utilized to monitor the electrochemical performance of the electrodes. Figure 5a, b shows the CV curves of all the samples at the scan rates of 50 and 200 mV s−1 in 6 mol L−1 KOH solution. All the CV curves are close to ideal rectangular shapes without any redox peaks in the potential range from 0 to 1.0 V, demonstrating the excellent capacitive behavior and rapid charging/discharging characteristics of these electrodes [22]. Galvanostatic charge/discharge curves of CAs, PCA-0.5, and PCA-1.0 electrodes at a current density of 1 A g−1 are shown in Fig. 6. All the charge/discharge curves exhibit a typical isosceles triangle distribution without any obvious decreases in IR, indicating good capacitive behaviors.

CV curves of samples obtained at scan rates of a 50 mV s−1 and b 200 mV s−1

Galvanostatic charge/discharge curves of samples at current density of 1 A g−1
The gravimetric charge/discharge specific capacitances are performed at various current densities from 0.05 to 100 A g−1 (Fig. 7a) to evaluate the rate properties of the electrodes. For all the samples, specific capacitances decrease along with the increase of current densities. When the current density is below 10 A g−1, the specific capacitances of all the electrodes decrease sharply due to the low rate of ion diffusion within micropores which is a common phenomenon for various carbon electrodes [8, 22], and the PCA-1.0 electrode possesses the highest specific capacitance due to the largest specific surface area. However, as the current density increases above 10 A g−1, the specific capacitance of PCA-0.5 becomes higher than PCA-1.0 instead. The specific capacitances of CAs, PCA-0.5, and PCA-1.0 reach 156.1, 227.9, and 261.2 F g−1 at a current density of 0.05 A g−1, and still remain 77, 175.4, and 145.5 F g−1 at a current density of 100 A g−1, respectively.

a Gravimetric specific capacitances of samples at different current densities and b cycle performance of samples at current density of 1 A g−1
Galvanostatic charge/discharge test for 5000 cycles at a current density of 1 A g−1 (Fig. 7b) was carried out to investigate the lifespan and durability of the samples. The as-prepared samples exhibit a capacitance retention ratio of 92.3 % (CA), 91.8 % (PCA-0.5), and 88.8 % (PCA-1.0) after 5000 cycles, respectively, demonstrating a good long-term electrochemical stability.
As a high-specific capacitance and a long cycling life are important requirements for supercapacitors applications [23], the encouraging results indicate that PCAs are promising electrode materials in aqueous electrolyte. In addition, such superior performances of PCAs are competitive with some other electrode materials using in symmetric supercapacitors [24], such as activated coconut shell [25] (C max = 192 F g−1, 61.3 % retention after 3000 cycles), activated walnut shell [26] (C max = 202.8 F g−1, 79.4 % retention after 1000 cycles), activated coal-tar pitch [27] (C max = 224 F g−1, 98.5 % retention after 1000 cycles), RF-based activated CAs [28] (C max = 245 F g−1), activated carbon nanotubes [29] (C max = 57 F g−1), activated carbon nano fibers [30] (C max = 255 F g−1, 92 % retention after 10,000 cycles), and graphene [31] (C max = 255 F g−1, 93 % retention after 1200 cycles).
Electrochemical impedance spectroscopy (EIS) tests were conducted to investigate the effects of frequency on the electrodes in detail, and typical Nyquist plots are given in Fig. 8a. Each of the Nyquist plot consists of a semicircle in the high-frequency region, a Warburg-type line (the slope of portion of the 45° curve) in the intermediate-frequency region, and a vertical line in the low-frequency region. The intersection between semicircle and X-axis in the high-frequency region represents the equivalent series resistance (ESR), which is related to the intrinsic resistance of the electrode and the contact resistance between the electrode and the current collector [32, 33]. The ESR values of CAs, PCA-0.5, and PCA-1.0 are only 0.32, 0.36, and 0.40 Ω, respectively. In the enlarged view inserted in Fig. 8a, PCA-1.0 shows an obvious Warburg line which demonstrates the frequency dependence of ion diffusion/transportation from the electrolyte to the surface of the porous electrode. Due to collapse of large mesopores and macropores resulted from violent activation, PCA-1.0 shows a larger ESR and a slower ion diffusion/transportation rate than CAs and PCA-0.5. In the low-frequency region, the nearly vertical lines are observed, indicating the pure capacitive behavior of all the samples.

a Nyquist plots for samples in aqueous solution and b the equivalent circuit model used to fit the EIS data
The equivalent circuit model used to fit the EIS data is provided in Fig. 8b. The equivalent circuit includes elements of R S (resistance from the electrolyte and the contact resistance between the electrode and the current collector), R F (resistance from faradaic charge transfer reaction), CPE1 (constant phase element for faradaic charge transfer reaction), W O (Warburg impedance), and CPE2 (constant phase element for electrochemical double layer). The impedance of CPE1 and CPE2 are determined by the following equation [34]:
where Z is impedance, T is capacitance of associated element, j is the imaginary unit (\( j=\sqrt{-1} \)), ω is angular frequency (ω = 2 π f), and α is a dimensionless parameter for fitting purpose.
Table 4 summarizes the fitting results of all the electrodes. As listed, the R S values are highly consistent with the values of ESR. Among all the samples, PCA-0.5 exhibits a relatively small W O value (Warburg impedance). The Warburg impedance is created by ion diffusion, which appears as a straight line with a slope of 45°. Therefore, the W O is mainly determined by the porous structure of the electrodes. With an appropriate KOH activation, PCA-0.5 maintains the 3D porous structure of CAs and introduces abundant micropores and large mesopores. The hierarchical porous structure of PCA-0.5 promotes the ion diffusion, which results in a smaller Warburg impedance than CAs and PCA-1.0. Due to the destruction of the porous structure by strong activation, PCA-1.0 exhibits an obvious Warburg-type line and a large W O value. R F represents the resistance from faradaic charge transfer reaction. PCAs are more disordered than CAs and contain a higher oxidized carbon content. The higher content of the oxidized carbon contributes to the larger R F value of PCAs.
Besides, Bode plots, the real capacitance (C′(ω)) and the imaginary capacitance (C″(ω)) of the complex capacitance (C(ω)) as a function of frequency, are provided for obtaining more insights into the electrochemical performances of the supercapacitors. The real capacitance (C′(ω)) as a function of frequency is shown in Fig. 9a. At the low frequency of 10 mHz, PCA-0.5 and PCA-1.0 present relatively high capacitance up to 500 and 640 F g−1, and the capacitance of CAs is only 390 F g−1, which highly accord with the results obtained from CV measurement. The capacitances of all the samples drop sharply with the increase of frequency in the region of 10 mHz to 10 Hz and tend to be less frequency dependent in the high-frequency region. Figure 9b presents the imaginary part of capacitance (C″(ω)) changes versus frequency. The C″(ω) goes through a maximum at a frequency f 0, defining a relaxation time constant (t 0) as t 0 = 1/f 0. The relaxation time constant is a characteristic of the whole system and can be a quantitative measurement for the speed of reversible charging/discharging process. The values of t 0 are only 0.8, 1.0, and 2.51 s for CAs, PCA-0.5, and PCA-1.0, respectively. The hierarchical porous structures of CAs and PCA-0.5, especially the existence of large mesopores and macropores, decrease the ion transfer resistance and distance, which contribute to the superior power output ability of the samples.

Bode plots of the real part (a) and imaginary part (b) of capacitance as functions of frequency
Next, the electrochemical performances of CAs, PCA-0.5, and PCA-1.0 electrodes are further discussed in organic electrolyte. Figure 10a presents the CV curves of the samples at the scan rate of 5 mV s−1 in 1 M Et4NBF4/PC solution. All the curves exhibit ideal rectangular-like shapes without obvious redox peaks in the potential region of 0 to 2.7 V, demonstrating the good reversibility of the electrodes in organic electrolyte. Besides, the specific capacitances of CAs, PCA-0.5, and PCA-1.0 calculated from galvanostatic charge/discharge curves at a low current density of 0.05 A g−1 (Fig. 10b) are 11.7, 128.4, and 155.4 F g−1, and still maintain 4.50 F g−1 (38.5 %), 111.5 F g−1 (86.8 %), and 140.3 F g−1 (90.3 %) as the current density increases to 1 A g−1, respectively. The results indicate the superior capacitive and rate performance of PCAs in organic electrolyte. Due to the diameter of Et4N+ ion (around 0.7 nm) in organic electrolyte is larger than the K+ ion (0.36 ∼ 0.42 nm) in the aqueous electrolyte, the diffusion of Et4N+ in micropores is more difficult than the diffusion of K+ [35]. Thus, CAs with few micropores and small mesopores exhibit relatively low specific capacitance and rate capacity in organic electrolyte. PCAs, especially PCA-1.0, maintain good capacitive and rate behaviors in organic electrolyte, which are mainly because of the abundance of interconnected micropores and small mesopores.

a CV curves of samples in 1 M Et4NBF4/PC electrolyte obtained at the scan rate of 5 mV s−1. b Gravimetric specific capacitance of samples versus cycle number at various current densities
The superior electrochemical performance of the PCA electrodes in both aqueous and organic electrolytes can be reasonably attributed to the unique architectures of PCAs. On one hand, the abundant micropores and small mesopores of PCAs provide a large and effective surface area for efficient electrical charge storage, which result in a high-specific capacitance. On the other hand, the adequate large mesopores and macropores which could act as ion-buffering reservoirs shorten the ion transportation path. Moreover, the hierarchical pore texture with micropores, mesopores, and macropores facilitate the transport of ions, ensuing high-power density and excellent rate capability.
Conclusion
We have successfully synthesized the ACM-based porous carbon aerogels by a facile method. The as-prepared PCAs exhibit hierarchical porous network and large specific area. Symmetric supercapacitors fabricated using PCAs as electrodes demonstrate superior gravimetric specific capacitance, high-rate capability, and excellent cycle performance in both aqueous and organic electrolytes. The intriguing results endow that PCAs are a new type of competitive electrode materials for supercapacitors. In addition, due to its interesting microstructures, PCAs may provide a promising material for the applications of hydrogen and electrical energy storage, desalination, and catalysis.
References
Zhang LL, Zhao XS (2009) Chem Soc Rev 38:2520–2531
Zhu Y, Murali S, Stoller MD, Ganesh KJ, Cai W, Ferreira PJ, Pirkle A, Wallace RM, Cychosz KA, Thommes M, Su D, Stach EA, Ruoff RS (2011) Science 332:1537–1541
Wang G, Zhang L, Zhang J (2012) Chem Soc Rev 41:797–828
Zhai Y, Dou Y, Zhao D, Fulvio PF, Mayes RT, Dai S (2011) Adv Mater 23:4828–4850
Lee SW, Gallant BM, Byon HR, Hammond PT, Shao-Horn Y (2011) Energy Environ Sci 4:1972–1985
Kotz R, Carlen M (2000) Electrochem Acta 45:2483–2498
Chen MD, Kang XY, Wumaier T, Dou JQ, Gao B, Han Y, Xu GQ, Liu ZQ, Zhang L (2012) J Solid State Electrochem 17:1005–1012
Lv YY, Zhang F, Dou YQ, Zhai YP, Wang JX, Liu HJ, Xia YY, Tu B, Zhao DY (2012) J Mater Chem 22:93–99
Jiang HL, Liu B, Lan YQ, Kuratani K, Akita T, Shioyama H, Zong F, Xu Q (2011) J Am Chem Soc 133:11854–11857
Chen Z, Weng D, Sohn H, Cai M, Lu YF (2012) RSC Adv 2:1755–1758
Liu D, Shen J, Liu NP, Yang HY, Du A (2013) Electrochim Acta 89:571–576
Pröbstle H, Wiener M, Fricke J (2003) J Porous Mater 10:213–222
Biener J, Stadermann M, Suss M, Worsley MA, Biener MM, Rose KA, Baumann TF (2011) Energy Environ Sci 4:656–667
Wang DW, Li F, Liu M, Lu GQ, Cheng HM (2008) Angew Chem Int Ed Engl 47:373–376
Halama A, Szubzda B, Pasciak G (2010) Electrochim Acta 55:7501–7505
Li J, Wang XY, Huang QH, Gamboa S, Sebastian PJ (2006) J Power Sources 158:784–788
Tateishi D, Esumi K, Honda H, Oda H (1992) Carbon 30:942–944
Wang J, Chen MM, Wang CY, Wang JZ, Zheng JM (2011) J Power Sources 196:550–558
Biniak S, Szymanski G, Siedlewski J, Swiatkowski A (1997) Carbon 35:1799–1810
Chen J, Yao BW, Li C, Shi GQ (2013) Carbon 64:225–229
Wang JB, Yang XQ, Wu DC, Fu R, Dresselhaus MS, Dresselhaus G (2008) J Power Sources 185:589–594
Yan J, Xiao Y, Ning GQ, Wei T, Fan ZJ (2013) RSC Adv 3:2566–2571
Hou Y, Cheng YW, Hobson T, Liu J (2010) Nano Lett 10:2727–2733
Wang JC, Kaskel S (2012) J Mater Chem 22:23710–23725
Mi J, Wang XR, Fan RJ, Qu WH, Li WC (2012) Energy Fuels 26:5321–5329
Choi WS, Shim WG, Ryu WD, Hwang MJ, Moon H (2012) Microporous Mesoporous Mater 155:274
He X, Li R, Qiu J, Xie K, Ling P, Yu M, Zhang X, Zheng M (2012) Carbon 50:4911
Wang J, Yang X, Wu D, Fu R, Dresselhaus MS, Dresselhaus G (2008) J Power Sources 185:589
Xu B, Wu F, Su Y, Cao G, Chen S, Zhou Z, Yang Y (2008) Electrochim Acta 53:7730
Barranco V, Lillo-Rodenas MA, Linares-Solano A, Oya A, Pico F, Ibanez J, Agullo-Rueda F, Amarilla JM, Rojo JM (2010) J Phys Chem 114:10302
Lei Z, Lu L, Zhao XS (2012) Energy Environ Sci 5:6391
Wang Q, Yan J, Wang YB, Ning GQ, Fan ZJ, Wei T, Cheng J, Zhang ML, Jing XY (2013) Carbon 52:209–218
An KH, Kim WS, Park YS, Moon JM, Bae DJ, Lim SC, Lee YS, Lee YH (2001) Adv Funct Mater 11:387–392
Liu X, Juan L, Zhan L, Tang L, Wang Y, Qiao W (2010) J Electroanal Chem 642:75–81
Ue M (1994) J Electrochem Soc 141:3336–3342
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 51372168) and the Key Project of Natural Science Foundation of Tianjin City of China (No. 12JCZDJC27000)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yuan, Y., Zhang, C., Wang, C. et al. Amphiphilic carbonaceous material-based hierarchical porous carbon aerogels for supercapacitors. J Solid State Electrochem 19, 619–627 (2015). https://doi.org/10.1007/s10008-014-2638-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-014-2638-1