
Abstract
Antibiotic resistance is threatening the medical industry in treating microbial infections. Many organisms are acquiring antibiotic resistance because of the continuous use of the same drug. Gram-negative organisms are developing multi-drug resistance properties (MDR) due to chromosomal level changes that occurred as a part of evolution or some intrinsic factors already present in the organism. Stenotrophomonas maltophilia falls under the category of multidrug-resistant organism. WHO has also urged to evaluate the scenario and develop new strategies for making this organism susceptible to otherwise resistant antibiotics. Using novel compounds as drugs can ameliorate the issue to some extent. The β-lactamase enzyme in the bacteria is responsible for inhibiting several drugs currently being used for treatment. This enzyme can be targeted to find an inhibitor that can inhibit the enzyme activity and make the organism susceptible to β-lactam antibiotics. Plants produce several secondary metabolites for their survival in adverse environments. Several phytoconstituents have antimicrobial properties and have been used in traditional medicine for a long time. The computational technologies can be exploited to find the best compound from many compounds. Virtual screening, molecular docking, and dynamic simulation methods are followed to get the best inhibitor for L1 β-lactamase. IMPPAT database is screened, and the top hit compounds are studied for ADMET properties. Finally, four compounds are selected to set for molecular dynamics simulation. After all the computational calculations, withanolide R is found to have a better binding and forms a stable complex with the protein. This compound can act as a potent natural inhibitor for L1 β-lactamase.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Antibiotics have played an imperative role in treating microbial infections and achieving amelioration in medicine and surgery [1]. Life expectancy has increased since the discovery and use of antibiotics because they act by altering the course of bacterial diseases [2][2]. However, the overuse of antibiotics aided in developing resistance in some microbes [4]. It was found that there is a link between antibiotic use and the establishment and spread of antibiotic-resistant bacterial strains (the antibiotic alarm 2013). Gene encoding antibiotic resistance can be inherited from relatives or nonrelatives on mobile genetic components like plasmids in bacteria. The horizontal gene transfer (HGT) mechanism can transfer antibiotic resistance between bacteria species. Mutation can also be the reason behind the rise in antibiotic resistance. Antibiotics can phase out drug-sensitive organisms, but the resistant ones that are naturally selected are left behind to multiply. Over prescription of antibiotics is one of the most important reasons behind the emergence of antibiotic resistance. Antibiotic resistance is one of the major problems concerning the health field worldwide. In India, over 56,000 newborns die yearly from sepsis caused by antibiotic-resistant organisms resistant to first-line antibiotics [5]. The antimicrobial resistance property of gram-negative bacteria (GNB) makes it cumbersome for physicians to treat ICU patients. 45–70% of ventilator-associated pneumonia cases (VAP) [6] and about 20–30% of catheter-related blood-infection cases are caused by GNB. ICU-acquired sepsis-like surgical site infections and UTIs are some common GNB infections among nosocomial patients in hospitals. To overcome the current antibacterial resistance menace, there is an escalated demand to discover novel antibiotics against those neglected targets (Worthington and Melander, 2013). Most antibiotics are attenuated by enzymatic hydrolysis or degradation; in some cases, non-enzymatic mechanisms contribute to antibiotic resistance [7]. Both enzymatic and non-enzymatic mechanisms of resistance can either be intrinsic (effect of some intrinsically present chromosome) or may be acquired over time as a result of mutation [8] [9]. Bacteria utilize mechanisms like efflux pump, reduced outer membrane permeability, drug target modification, and drug inactivation by β-lactamase enzymes to attenuate the drug effects [10].
β-lactamases are a group of enzymes capable of hydrolyzing the amide bond in the β-lactam ring of β-lactam antibiotics such as carbapenems, penicillin and cephalosporin, and monobactam [11]. The two families of β-lactamases include “Metallo-β-lactamase” and “Serine-β-lactamases” [12]. Serine-based β-lactamases belong to class A, class C, and class D. conversely, class B enzymes bind one or two metal ions, usually Zn2+, and play a crucial part in their catalytic activity earning them the name metallo-lactamases (MBL). The effect of metal ion binding to the active site is found to enhance the catalytic significance of the enzyme. In the case of São Paolo metallo-β-lactamase-1, the presence of two zinc ions at the active site provides a closed conformation and enhances the catalytic property. Meanwhile, binding of one or no zinc gives an open conformation to the active site [13]. Similarly, the double zinc coordination at the active site of New-Delhi metallo-β-lactamase 1 is necessary for binding antibiotics (ampicillin). Also, it restricts the motion in the loop region [14]. Based upon the substrate profile, protein sequence, and slight changes in the active site configuration for two Zn.2+ ions, MBL proteins are divided into three subclasses: B1, B2, and B3. MBLs belonging to the B1 and B3 classes can hydrolyze practically every antibiotic with β-lactam rings, including the most recently produced carbapenems. Class B2 enzymes, on the other hand, have a narrow carbapenem substrate profile and weak action against penicillin and cephalosporins [15]. L1-β-lactamases belong to the MBL family, while L2-β-lactamase belongs to the serine β-lactamase family [16]. Multiple biochemical studies support several crystal structures of L1 and other B3 MBLs complexed with antibiotics [17][17]. Even though the B3 MBLs are structurally well-conserved, especially at the active site flanked with two zinc ions, the sequence identity is 23–35% [17][17]. Inhibitors for β-lactamase are prioritized to aggrandize treatment potency with β-lactam antimicrobials and avoid antimicrobial resistance. β-lactamase inhibitors may act as “competitive inhibitors” or “suicide inhibitors” that can permanently render the enzyme inoperable utilizing secondary chemical reactions in the active site. Clavulanic acid, a natural product discovered in 1976 and used with ticarcillin and amoxicillin, is the first clinically successful β-lactamase inhibitor. It was quickly followed by introducing sulbactam and tazobactam, synthetic penicillin-based sulfone-lactamase inhibitors combined with ampicillin and piperacillin. The β-lactamase inhibitors that are clinically used for treatment like clavulanic acid have now become ineffective [20]
Pathogens encompassing β-lactamase enzymes have sprung up in clinical fields and the environment. Stenotrophomonas maltophilia is an emerging prokaryotic, gram-negative, nosocomial, multi-drug resistant non-fermentative bacterium. The organism has emerged as an important hospital-acquired pathogen over the past decade. Treatment of infections due to this bacterium is challenging due to its multi-drug resistance (MDR). MDR is the property or ability of an organism to resist the effect of at least three antibiotics. The MDR in S.maltophilia is due to the intrinsic multidrug resistance phenotype, which involves the impact of several chromosomal determinants. S.maltophilia expresses specialized efflux pumps that can pump out a broad spectrum of drugs, especially β-lactams. One among them, SmeDEF, is the most effective one and is efficient in the expulsion of quinolones and aminoglycosides [21]. The L1 β-lactamases or Zn β-lactamases and L2 β-lactamases with serine in the active site are the two chromosomal β-lactamases produced by S.maltophilia [22]. The L1 in S.maltophilia belongs to the B3 subclass of MBL [23]. The protein is a tetramer with αβ/βα folds. The active site is found between the two β sheets containing two zinc atoms, and the side chains that coordinate the metal ions are at the bottom of the active site. The amino acids bound to Zn1 are His196, His118, and His116 in 2.1–2.2 Å distance range. Asp120, His263, and His121 are bound to Zn2 with distances of 3.0, 2.5, and 2.1 Å, respectively [24]. An increase in the inhibitory capacity of L1 β-lactamase can be attributed to the two zinc ions present at the active site of the enzyme. Zinc plays a catalytic role in the enzyme. It stabilizes the tetrahedral transition state, protonates the nitrogen atom in the β-lactam ring, and causes a nucleophilic attack on the hydroxide group in the carbonyl carbon, thereby forming a tetrahedral intermediate [25][25]. The ligands mainly bind to the metal ion. The binding pocket of the protein provides a broader region for many β-lactam antibiotics to bind [24]. It has thus become an increasing concern to find inhibitors against β-lactamases that can render the enzyme ineffective and make the organism more susceptible to antibiotics.
Plants provide a vast range of medications used to treat several ailments. Many laboratories continue to screen medicinal plants for new antimicrobial medication candidates that can slow pathogen development or kill without causing toxicity to the host [27][27]. Plant-based antimicrobials hold significant therapeutic potential, making them a massive untapped supply of pharmaceuticals. They are found to help treat infectious disorders by avoiding most of the adverse side effects that are usually common when synthetic antibiotics are used for treatment. Plant-derived antimicrobials are biodegradable and are relatively safer drugs. Even though many antibiotics are available, the search for plant-based compounds to combat fungal and bacterial infections will continue [29].
In the current study, we screened the phytochemicals from IMPPAT database [30] to find an inhibitor against L1 β-lactamase of Stenotrophomonas maltophilia. After molecular docking, the compounds having good binding energy were selected for ADME and PAINS-remover studies (toxicity analysis). The compounds that passed the toxicity studies were set to molecular dynamic (MD) simulation to test the stability of the complex. Thus, the results will give a novel inhibitor identified for L1 β-lactamase of S.maltophilia.
Materials and methods
Virtual screening
Protein structure preparation
The X-ray crystallography structure of Metallo β-lactamase L1 from Stenotrophomonas maltophilia with hydrolyzed imipenem complex structure (PDB id: 6UAF [24]) was downloaded from the Protein Data Bank (RCSB-PDB) [25][25] and utilized for ensuing studies (Fig. 1). This protein structure resolution is 1.90 Å. The sequence length of Metallo-β-lactamase L1 is 293, and the downloaded crystal protein structure has 266 amino acids (residue number 23 to 289) with two Zn ions. However, missing residues present at the N-terminal and C-terminal were unresolved and these unsolved residues are not affecting the binding interaction as they are away from the active site. The protein structure was prepared by adding hydrogen and subjected to energy minimization through UCSF-Chimera software. For refining the structure and minimizing the energy of the selected protein, UCSF-Chimera software was employed.

Metallo-beta-lactamase L1 from Stenotrophomonas maltophilia protein (PDB ID: 6UAF) downloaded from Protein Data
Ligand library preparation
The ligand library for virtual screening of suitable inhibitor molecules against L1 β-lactamase was downloaded from the IMPPAT database [32]. The three-dimensional (3D) structures of the phytochemicals were downloaded in PDB format and then converted to PDBQT using Open Babel. The phytochemicals for which there was no availability of 3D structure were excluded from the study. A total of 7937 compounds were collected from the IMPPAT database.
Molecular docking
Molecular docking techniques are of great importance in the discovery of novel drugs. The methodology involves the prediction of the pose, virtual screening, and estimation of binding affinity of the ligands to the protein. The structure-based screening was performed using AutoDock Vina [33]. The protein in PDB format was converted to PDBQT format using Open Babel software [34]. Kollman’s charges method was utilized to add polar hydrogen and charges. The grid point in XYZ, 46 Å × 62 Å × 82 Å (x, y, and z), and the grid box center 20.245 × 24.393 × 0.68, with 0.375 Ǻ spacing, were assigned to the protein. The grid map was calculated using Autogrid4. The remaining docking computation parameters were maintained unchanged. After docking, a total of 10 conformations were taken into consideration for every ligand. A python script (https://vina.scripps.edu/manual/) was run to obtain the compounds with a high dock score (minimum binding energy). The interaction between protein–ligand complexes was calculated using PLIP [35]. A standard β-lactam drug, imipenem, was docked with the protein as a control.
ADME AND PAINS analysis
After molecular docking, we performed Absorption, Distribution, Metabolism, and Elimination (ADME) and PAINS-remover calculation to verify the pharmacokinetic ability of the selected compounds obtained after screening based on molecular docking. Lipinski’s rule, Ghose filter, Verber’s rule, Muegge’s rule, Egan’s rule, water solubility, lipophilicity, hepatotoxicity, etc. are some of the main drug-likeness rules. Along with the ADME calculation, we performed the PAINS (pan-assay interference compounds) calculation. Through this calculation, we removed the compounds that may give false-positive compounds results when predicting binding sites. Compounds with ADME and PAINS alerts must be eliminated from the study. For the calculation of ADME, we used the Swiss-ADME and for calculation of PAINS, we used PAINS-remover web online tools.
Molecular dynamics (MD) simulation
For performing MD, we used GROMACS (Version-4.6.5) software. GROMACS 53a6 force field was selected for generating the protein topology file. The binding orientation of the selected hit compounds was obtained from the docking site and the ligand topology was created using the PRODRUG online tool. For solvation, the complex was placed in a triclinic box and solvated with spc216 (simple point charge) water. Na + or Cl-ions were then added to the system for neutralization. The overall system (ions, water, and protein–ligand complex), was relaxed by running energy minimization with the steepest descent. PME algorithms were used for estimating electrostatic interaction. For this study, we set the pme_order to 4, fourier spacing to 0.16, and scale at 310 K. Next, the entire system was specified for thermal equilibration (NVT step) for 1 ns using modified Berendsen thermostat (V-rev100ns for selected virtual hit compounds). NPT was performed at 300 K using V-rescale, the pressure coupling (P-coupling), and temperature coupling (T-coupling). 100 ns MD simulation was performed without position restraints. The output was saved every 2 ps. At every 10 ps, the velocities and coordinates, nstvout and nstxout, respectively, were also held. We set the 1.0 mm cut-offs for coulombic (rcoulomb) and Van der Waals force (rvdw). Particle mesh Ewald (PME) and periodic boundary conditions (PBC) were used in 100 ns MD simulation to calculate the long-range electrostatic interactions. MD simulation results were analyzed by plotting the graph of root mean square deviation (RMSD), root mean square fluctuation (RMSF), and radius of gyration (Rg). They were calculated using g_rms, g_rmsf, and g_gyrate. Along with this, we checked the changes in the secondary structure of protein using the do_dssp program.
Binding free energy calculation using MM-PBSA
Once we finish the 100 ns Simulation, we use the stable region to calculate the binding free energy (BFE). Binding free energy calculation is a critical approach to study the reciprocal recognition and binding of protein and their ligands. The negative and positive values of free energy show the possibility of a reaction, and the importance of binding free energy is an accurate standard for evaluating the bending degree of protein receptors to ligands [36]. The binding free energy for selected hit compounds and the energy change under vacuum were calculated using molecular mechanics energies combined with Poisson-Boltzmann (MM-PBSA) method by g_mmpbsa Tool [37]. PB shows the polar part of solvent-free energy of systems calculated by the Poisson-Boltzmann (PB) equation. The non-polar part of solvent-free energy systems is fitted by the solvent-accessible surface area (SASA). The MMMPBSA calculation is as follows \(: Increment\Delta Gbind=\Delta Evdw+ \Delta Eele+\Delta Gpol+\Delta Gnonpol-T\Delta S\)
The ΔEvdW and ΔEele are van der Waals and electrostatic components, respectively, and the polar and nonpolar components are indicated as ΔGpol and ΔGnonpol, respectively. TΔS is the temperature and entropic contribution towards BFE. BFE plays a significant role in drug discovery, giving a quantitative estimation of the ligand binding to the protein. The q_mmpbsa tool calculated MM-PBSA after extracting the stable segment in the trajectory file.
Results and discussion
Virtual screening
Structure-based virtual screening was used to screen down the compounds downloaded from Indian Medicinal Plants, Phytochemistry and Therapeutics (IMPPAT) database using molecular docking techniques. Molecular docking was performed using standard default.
parameter to calculate the binding energy. A total of 7937 IMPPAT phytochemical compounds were docked with the target protein (6uaf), and compounds with the best dock score (more negative value) were considered to be the best bonding. Out of 7937 phytochemicals, we selected the top eight compounds based on their minimum binding energy from 10 conformations generated by all the compounds. The selected eight compounds are withanolide R (− 10.26 kcal/mol), withanolide A (− 9.59 kcal/mol), 27-Deoxywithaferin (− 9.98 kcal/mol), demissidine (− 9.3 kcal/mol), pibenzimol (− 9.3 kcal/mol), crinasiatine (− 9.2 kcal/mol), 3-Methylcholanthrene (− 8.7 kcal/mol), and withanolide Q (− 6.14 kcal/mol). These selected eight compounds were further screened down by applying the ADME and PAINS parameter to get the good drug compound (Tables 1 and 2).
ADME and PAINS analysis
All the top eight compounds, after docking, were studied for ADMET and PAINS properties. All the compounds had a molecular weight of less than 500 Daltons and logP < 5. One violation of the Ghose rule occurs for all the compounds except for 3-Methylcholanthrene (− 8.7). One of the selected compounds, demissidine, with a dock score of − 9.3, violated one Lipinski’s rule and one Muegge’s rule. 3-Methylcholanthrene violated one Lipinski’s rule and two Muegge’s rules. All the compounds were AMES non-toxic. The ADME study results are given in Table 3. After ADME calculation, we run the PAINS-remover to check if the compounds represent poor choices for drug development. None of the compounds showed PAINS alert. After the ADME and PAINS analysis, we selected four compounds which showed no PAINS alert, satisfied most of the drug-likeness parameters, are AMES non-toxic and have very low CYP p450 inhibition. The selected compounds are withanolide Q, withanolide A, withanolide R, and 27-Deoxywithaferin. These compounds are present in and can be extracted from, the roots of Withania somnifera (ashwagandha). These selected four compounds were finalized to proceed for the MD simulation.
Molecular docking analysis
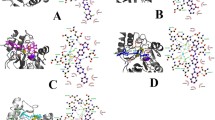
Based on binding energy, ADME, and PAINS analysis we select the four compounds. 25-Deoxywithaferin, withanolide A, withanolide Q, and withanolide R compounds interaction were calculated using the PLIP online tool. It shows the hydrogen bond and hydrophobic bond formed with protein and ligand, as well as it shows the interaction between protein–ligand complex with two zinc atoms present in the binding pocket. Further, we included Imipenem, the known standard reference compound (control) for comparison. It is an intravenous β-lactam antibiotic used for the treatment of infections. All these chosen compounds bind in the same declaration region near Zn ions, as shown in Figs. 1, 2, 3, 4, 5. The binding energies of these four compounds and the control are shown in Table 1. When compared to other selected hit compounds, withanolide R shows the most potent binding energy which is − 10.26 kcal/mo. There are two Zn ions present in L1 β-lactamase that interact with protein as well as the ligand. Compound withanolide A and withanolide R take part in interaction with Zn (Figs. 4 and 6) and the other two compounds withanolide Q and 27-Deoxywithaferin do not interact with Zn ions (Figs. 3 and 5). The amino acids involved in the formation of hydrogen bonds, hydrophobic interaction, salt bridge formation as well as interactions with Zn ions are mentioned in Table 1. For protein and 27-Deoxywithaferin complex, 302 Zn ion interactions are established with Asp109 (2.8 Å) and His110 (2.0 Å) and 303 Zn ion interact with His105 and His181 at 2.0 Å (Fig. 3A and B) (Table 1). Compound 27-Deoxywithaferin shows the hydrophobic interaction with Leu59, Phe145, Ile149, His246, and Lys277 (Fig. 3C). In the case of withanolide A-protein complex, Zn ion shows the interaction with amino acids and ligand shown in Fig. 4A and B (Table 1). This complex shows the hydrogen and hydrophobic interaction with Leu207, Ser208, Lys277, and Tyr32, and hydrophobic interaction with Trp38, Phe145, His246, Ala249, Tyr270, and Ala274 (Fig. 4C). Along with these interactions we observe the salt bridge formation at His105, His107, His110, His181, and His246 (Table 1

Protein–ligand complex formed by selected hit compounds. All selected hit compounds bond in the same place bear to two Zn atoms (Red color)

Interaction between protein–ligand (yellow color) and Zn ions (green color). A Zn ion 302 showing interaction with protein amino acids. B Zn ion 303 showing

Interaction between protein–ligand and Zn ions. A Withanolidine-A and Zn ion 302 showing interaction with protein amino acids. B Withanolidine-A and Zn ion 303

Interaction between protein–ligand and Zn ions. A Zn ion 302 showing interaction with protein amino acids. B Zn ion 303 showing interaction with protein

Interaction between protein–ligand and Zn ions. A Withanolidine-R and Zn ion 302 showing interaction with protein amino acids. B Withanolidine-R and Zn ion 303
and Fig. 4C). From the selected compounds, withanolide Q-protein complex, Zn ions do not show any interactions with the ligand, but form bonds with amino acids in the vicinity. The Zn302 interacts with His109, His110, and His246 at distance 2.1 Å, 2.2 Å, and 2.1 Å respectively and Zn303 interact with His105 (2.0 Å), His107 (2.2 Å), and His181 (2.2 Å) as shown in Fig. 5A and B and Table 1. This compound takes part in hydrogen and hydrophobic interaction with protein as shown in Table 1 and Fig. 5C. Withanolide R shows the interaction with Zn ions. Both Zn ion forms interaction with the compound as well as with amino acids as shown in Fig. 6AAand B and Table 1. Hydrogen bonds interaction were formed with Leu207, Ser208, His246, and Lys277, and hydrophobic interaction were established with His107, Phe145, Ile149, His246, Ala249, Tyr270, Ala274, and Lys277 residues (Fig. 6C and Table 1). Salt bridge interaction with His105, His107, His110, His181, and His246 is also established (Fig. 6C and Table 1). Among all four compounds, 27-Deoxywithaferin is not involved in forming hydrogen bonds with the protein, or any kind of bond is formed with Zn ions. The control (Imipenem) formed hydrogen bonds with amino acids Tyr32, Ser206, Lue207, and Ser208. Hydrophobic bond is formed with Ala249. It takes part in interaction with both Zn ions. This comparison analysis shows that the known β lactam drug (imipenem) and hit compound have the same pattern of interaction as well as hit compounds binds in the same region where the β lactam drug binds.
Molecular dynamics (MD) simulation analysis
Selected hit compounds were further subjected to 100 ns MD simulation to analyze the stability of the protein–ligand complex. Through this, we investigated the binding mechanism and dynamic behavior of the complexes. The receptor-ligand conformational stability was studied through RMSD, RMSF, and Radius of gyration. We calculated the hydrogen bond between protein–ligand and protein solvent throughout the 100 ns MD simulation. We checked the changes in the secondary structure when selected compounds were bound to the protein. After that, MMPBSA was computed to calculate binding free energy.
We calculated the RMSD value to check the change in the α-carbon of the ligand-receptor complex. An elevation of RMSD value at the starting stage of MD simulation up to 0.35 nm was observed for 27-Deoxywithaferin and withanolide R. withanolide A and withanolide Q showed the most RMSD value (0.45 nm), and it gradually increased up to 15 ns simulation. 27-Deoxywithaferin showed a slight increase in the RMSD value at the initial stage up to 1 ns and then there was a sudden decrease in it. After that, the RMSD value slowly increased up to 10 ns. Then the RMSD became constant with slight fluctuation throughout the 100 ns simulation between 0.4–0.5 nm (Fig. 7). In the case of withanolide R, there was more oscillation in value up to 5 ns, after that, it showed an increase in the RMSD value (0.55 nm) up to 15 ns. Up to 43 ns, the RMSD remained almost constant. There was a sudden decrease in RMSD between 0.40 and 0.55 nm up to 57 ns and then it gradually increased up to 100 ns MD simulation. In the case of withanolide A and withanolide Q, the same pattern of fluctuation was observed up to 35 ns simulation. After that Withanolide A showed a decreased RMSD value between 0.45 and 0.55 nm and withanolide-Q showed a slight increase between 0.55 and 0.60 nm throughout the 100 ns simulation (Fig. 7).

The RMSD for the virtual hit four compounds interact with protein and forms a complex
Further, we checked the momentum of each residue throughout the simulation when the ligand binds to the protein in the presence of Zn ions. For that, the RMSF was plotted against residues (Fig. 8). The plot shows that all compounds show the same protein fluctuation pattern. However, when withanolide R binds to protein, the initial amino acid shows a more RMSF value, and other amino acids indicate the same fluctuation pattern. Residues 210–230 showed a slight difference in RMSF value. In the case of withanolide Q, the binding to protein shows the same pattern of fluctuation by all amino acids, but residues 87–100 show a more RMSF value when compared with others. When withanolide A binds to the protein, all amino acids show a smaller RMSF value than others. 27-Deoxywithaferin and the protein complex shows the same fluctuation pattern throughout the simulation, but some amino acids show a lower RMSF value (140–150 residues and 210–223 residues). RMSF is more at residues from 245 to 260 (Fig. 8). This increase and decrease in the fluctuation are possibly due to interaction between the hit compound and the protein helix, small β-sheets, and the loop region of the protein. The RMSF plots backbone shows more fluctuation in the loop and helix regions.

The RMSF for the virtual hit compound interacts with protein and forms a complex
The compactness level of the protein–ligand complex in the presence of Zn ions was checked by calculating the radius of the gyration (Rg) plot (Fig. 9). We used the 100 ns MD simulation trajectory file for this calculation and plotted the gyration plot using Rg vs. time shown in Fig. 9. All selected hit compounds initially showed the Rg value between 1.82 and 1.85 nm. Then it decreased till 5 ns. Withanolide Q and withanolide R showed a slight increase in the Rg value up to 6 ns, and after that, the same pattern of fluctuation till 25 ns was observed. In the case of 27-Deoxywithaferin, the Rg value had a steady fluctuation. For withanolide A, the Rg value increased to 7 ns and then decreased to 20 ns. After some time, there was a sudden increase in Rg value (1.9 nm Rg). After this, the Rg decreased till 50 ns, and then there was a steady increase and decrease till the end of 100 ns simulation. At 45 ns, compounds 27-Deoxywithaferin and withanolide A showed the same pattern of Rg, which was between 1.78 and 1.83 nm. At the same point (45 ns), withanolide Q and withanolide R showed a slow increase in Rg value, and both compounds showed the same pattern of fluctuation up to 100 ns simulation.

The radius of gyration for the virtual hit four compounds interact with protein and form a complex
The H-bond interaction between protein–ligand and protein-solvent was also calculated (Figs. 10 and 11). Withanolide R showed a better hydrogen bond interaction than the other compounds. This compound formed 0–6 hydrogen bonds in 100 ns MD simulation. At initial simulation time up to 45 ns, 0–2 hydrogen bonds were established in the protein–ligand complex. At 73–92 ns, the number of hydrogen bonds increased to 2. 27-Deoxywithaferin (magenta color), withanolide A (purple color), and withanolide Q (green color) showed the same pattern of hydrogen bond formation Fig. 10. The H-bond interactions formed between the protein and the solvent were also calculated. 420–540 H-bonds were established between the protein and solvent (Fig. 11). Withanolide R had the highest number of hydrogen bond interactions among all the other compounds, and withanolide Q had the least. In the case of 27-Deoxywithaferin and withanolide A, the same pattern of hydrogen bond interactions between protein-solvent was observed.

Hydrogen bond interaction between protein–ligand complex formed throughout 100 ns MD simulation

Hydrogen bond interaction between protein-solvent formed throughout 100 ns MD simulation
Throughout the simulation, secondary structure changes were analyzed using the do_dssp program (Fig. 12). For this program, the trajectory file for 100 ns was used to compute the secondary structure for every frame. When the complexes are formed with the protein by selected hit compounds, some changes in the conformational behavior and degree of protein folding were observed. This depended on its secondary structure. When we compared selected hit compounds bound to the protein, no more secondary structural changes were observed. We observed some minor changes in the secondary structure, which is observed in β-sheets (red color) to turn (yellow color), coil (white color) to turn (yellow color), and turn (yellow color) to bend (green color).

Secondary structural conformation changes of hit compounds were shown during 100 ns simulation
Further, we calculated the binding free energy for the selected hit compounds (Table 4). Among the selected hit compounds, withanolide R showed the most binding free energy, − 80.003 kJ/mol, and compound 27-Deoxywithaferin showed the least binding free energy, − 28.096 kJ/mol. The other two compounds binding energy are shown in Table 4. The MMPBSA energy is known as approximate free energies of binding. Stronger critical interaction gives more negative values.
Discussion
Stenotrophomonas maltophilia, a multidrug-resistant organism, makes treating the infections caused by the bacteria very difficult. This organism can resist several antibiotics that are currently prescribed for microbial infections. Some antibiotics like fluoroquinolones (FQs), trimethoprim-sulfamethoxazole (SXT), ticarcillin clavulanate, tetracyclines, and ceftazidime are found to have in vitro activity against this bacteria; however, the clinical evidence for their use is limited [38][38] WHO has also urged on to come up with novel strategies to make the organism vulnerable to the antibiotics available. The presence of β-lactamase enzymes in the extracellular space makes the organism resistant to a broad spectrum of β-lactam antibiotics. There are no specific inhibitors discovered that inhibit the L1 β-lactamase. The active site of this enzyme contains two zinc ions at 302 (Zn1) and 303 (Zn2). Targeting the active site zinc ions will help inhibit the catalytic property of the protein and thus can lead to the discovery of a novel inhibitor specifically for L1 β-lactamase that can compete with β-lactam antibiotics to bind at the active site.
An in-silico approach toward discovering novel inhibitor compounds is less expensive and recommendable in today’s scenario. Many research data suggest that molecular docking and dynamics approaches in studying protein–ligand stability are effective [40][40]. With the development of molecular dynamics simulation techniques in the 1970s, the usage of the computational approach got elevated in drug discovery [42][42][42]. In the present study, a virtual screening, molecular docking, and MD simulation approach is carried out on plant-based compounds to screen the inhibitor for L1 β-lactamase from the 7937 phytochemicals collected from the IMPPAT database. The docking results were compared with that of the reference compound (imipenem). It is a standard β-lactam antibiotic compound discovered by merck scientist Burton Christensen, William Leanza, and Kenneth Wildonger in the mid-1970s. L1 β-lactamase mainly inhibits imipenem. The selected hit metabolites had better dock score and binding interactions than imipenem, suggesting that they can compete with the β-lactam antibiotics (substrates for β-lactamase) and bind to the active site and block it, thereby reducing the ability of the enzyme in hydrolysis of the antibiotics. Withanolide A, withanolide Q, withanolide R, and 27-Deoxywithaferin were selected by molecular docking, ADME, and PAINS-remover studies. All the four compounds are derivatives of withanolides in Withania somnifera (ashwagandha). Most of the pharmacological properties of W.somnifera are rendered by withanolides. They have many biological activities, including immunoregulatory, anticancer, antimicrobial, anti-inflammatory, leishmanicidal, and trypanocidal [45]. Withanolide A has suitable neuropharmacological activities that aid in the outgrowth of the neurite. Thus it can reverse neuritic atrophy and help to reconstruct the synapse [46]. Withanolide Q is reported to have a modulating effect on several SARS-CoV-2 proteins [47]. 27-Deoxywithaferin is found to have antibacterial properties [48] and has the potential to inhibit spike protein of SARS-CoV-2, while withanolide R can inhibit the main protease of SARS-CoV-2 [49]. These data suggest the strong antimicrobial property of these phytochemicals.
27-Deoxywithaferin (BE − 9.98 kcal/mol) showed a stable and lower RMSD value than all the other compounds, and thereby it is understood that this complex shows a minimal deviation in the path from the actual when compared to other complexes. On the other hand, RMSF result also shows the same pattern of fluctuation throughout the simulation. Comparison of the RMSF of the hit compounds suggest that all these compounds binding to the zinc-containing L1 β-lactamase do not show much difference and even the amino acids present in the active site do not show much fluctuation. There were fluctuations at some residues, but none of these residues are present in the loop region, and hence it did not affect the stability of the complexes during the 100 ns simulation. Even though 27-Deoxywithaferin had a good RMSD and RMSF, the compound did not form any hydrogen bonds with the protein, which resulted in the least binding energy, and it did not interact with zinc atoms at the active site. To study the compactness of the complex formed between protein and selected hit compounds in the presence of Zn ions, the Rg plot was analyzed. From the Rg plot, it can be elucidated that the compactness level of the 27-Deoxywithaferin protein complex was stable and constant throughout the simulation, but at the initial time period it showed a decrease in the compactness up to 5 ns but after 5 ns its shows the steady and stable compactness compared to the other three complexes. Withanolide A showed the least compactness. Withanolide Q and R showed the same pattern of compactness as their Rg was stable throughout the 100 ns simulation. We can get an idea about the binding strength by analyzing the number of H-bonds established between the compounds, and the number of hydrogen bonds based between the compounds gives an idea about the strength of binding.
Withanolide R established the most significant number of hydrogen bonds (between 0 and 6) with the residues at the active site of the target protein. This compound also had a better hydrogen bond interaction with the solvent molecules. Among the four compounds, withanolide R showed the slightest fluctuation which signifies the stability of the complex, and the MMPBSA score was maximum (− 80.003 kJ/mol). This can be because of its strong binding to the residues and zinc ions at the active site. All the data thus obtained based on computational calculations can give ideas for designing a potent drug that can specifically target the L1 β-lactamase and inhibit its action.
Conclusion
We successfully screened the phytochemicals from the IMPPAT database using molecular docking, ADME, and PAINS remover techniques. Good dock scores were obtained for ligands interacting with the amino acid residues and zinc ions at the active site. The ADME and PAINS-remove study on the top hit compounds resulted in four phytochemicals that satisfied the drug-likeness and lead likeness properties. We also carried out an MD simulation to find the most stable complex. From the entire study, the compound withanolide R present in Withania somnifera (ashwagandha) formed a stable binding conformation at the active site. It had the best dock score, binding energy, and the highest number of interactions with the active site residues. The conformational fluctuations were also less. The highest number of interactions at the active site and stability in binding can make the compound a potent inhibitor of the enzyme. However, in vitro studies have to be conducted to thoroughly understand and thereby confirm the inhibitory property of withanolide R on L1 β-lactamase of S.maltophilia.
Data availability
Not applicable.
References
Gould IM, Bal AM (2013) New antibiotic agents in the pipeline and how they can help overcome microbial resistance. Virulence 4:185–191. https://doi.org/10.4161/viru.22507
Rossolini GM, Arena F, Pecile P, Pollini S (2014) Update on the antibiotic resistance crisis. Curr Opin Pharmacol 18:56–60
Piddock LJV (2012) The crisis of no new antibiotics-what is the way forward? Lancet Infect. Dis 12:249–253
Read AF, Woods RJ (2014) Antibiotic resistance management. Evol Med Public Heal 2014:147. https://doi.org/10.1093/emph/eou024
Wattal C, Kler N, Oberoi JK et al (2020) Neonatal sepsis: mortality and morbidity in neonatal sepsis due to multidrug-resistant (MDR) organisms: part 1. Indian J Pediatr 87:117–121
Barbier F, Andremont A, Wolff M, Bouadma L (2013) Hospital-acquired pneumonia and ventilator-associated pneumonia: recent advances in epidemiology and management. Curr Opin Pulm Med 19:216–228
Alekshun MN, Levy SB (2007) molecular mechanisms of antibacterial multidrug resistance. Cell 128:1037–1050
Luyt CE, Bréchot N, Trouillet JL, Chastre J (2014) Antibiotic stewardship in the intensive care unit. Crit Care 18: https://doi.org/10.1186/s13054-014-0480-6
Bassetti M, De Waele JJ, Eggimann P et al (2015) Preventive and therapeutic strategies in critically ill patients with highly resistant bacteria. Intensive Care Med 41:776–795
Kapoor G, Saigal S, Elongavan A (2017) Action and resistance mechanisms of antibiotics: a guide for clinicians. J Anaesthesiol Clin Pharmacol 33:300–305
Hamed RB, Gomez-Castellanos JR, Henry L et al (2013) The enzymes of β-lactam biosynthesis. Nat Prod Rep 30:21–107
Rahman M, Khan MKA (2020) In silico based unraveling of New Delhi metallo-β-lactamase (NDM-1) inhibitors from natural compounds: a molecular docking and molecular dynamics simulation study. J Biomol Struct Dyn 38:2093–2103. https://doi.org/10.1080/07391102.2019.1627248
Chen J, Wang J, Pang L et al (2021) Deciphering molecular mechanism behind conformational change of the São Paolo metallo-β-lactamase 1 by using enhanced sampling. J Biomol Struct Dyn 39:140–151. https://doi.org/10.1080/07391102.2019.1707121
Chen J, Wang J, Physics WZ-PCC, 2017 undefined Zinc ion-induced conformational changes in new Delphi metallo-β-lactamase 1 probed by molecular dynamics simulations and umbrella sampling. pubs.rsc.org
Palzkill T (2013) Metallo-β-lactamase structure and function. Ann N Y Acad Sci 1277:91–104. https://doi.org/10.1111/j.1749-6632.2012.06796.x
Tooke CL, Hinchliffe P, Lang PA, et al (2019) Molecular basis of class A β-lactamase inhibition by relebactam. Antimicrob Agents Chemother 63: https://doi.org/10.1128/AAC.00564-19
Nauton L, Kahn R, Garau G et al (2008) Structural insights into the design of inhibitors for the L1 metallo-β-lactamase from stenotrophomonas maltophilia. J Mol Biol 375:257–269. https://doi.org/10.1016/j.jmb.2007.10.036
Crisp J, Conners R, Garrity JD et al (2007) Structural basis for the role of Asp-120 in metallo-β-lactamases. Biochemistry 46:10664–10674. https://doi.org/10.1021/bi700707u
Wachino JI, Yamaguchi Y, Mori S et al (2013) Structural insights into the subclass B3 metallo-β-lactamase SMB-1 and the mode of inhibition by the common metallo-β-lactamase inhibitor mercaptoacetate. Antimicrob Agents Chemother 57:101–109. https://doi.org/10.1128/AAC.01264-12
Chatwin CL, Hamrick JC, Trout REL, et al (2021) Microbiological characterization of VNRX-5236, a broad-spectrum b-lactamase inhibitor for rescue of the orally bioavailable cephalosporin ceftibuten as a carbapenem-sparing agent against strains of enterobacterales expressing extended-spectrum b-lactamases and serine carbapenemases. Antimicrob Agents Chemother 65: https://doi.org/10.1128/AAC.00552-21/SUPPL_FILE/AAC00552-21_SUPP_1_SEQ1.PDF
Brooke JS (2012) Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin Microbiol Rev 25:2–41
Looney WJ, Narita M, Mühlemann K (2009) Stenotrophomonas maltophilia: an emerging opportunist human pathogen. Lancet Infect Dis 9:312–323
Juan C, Torrens G, González-Nicolau M, Oliver A (2017) Diversity and regulation of intrinsic β-lactamases from non-fermenting and other Gram-negative opportunistic pathogens. FEMS Microbiol Rev 41:781–815
Kim Y, Maltseva N, Wilamowski M et al (2020) Structural and biochemical analysis of the metallo-β-lactamase L1 from emerging pathogen stenotrophomonas maltophilia revealed the subtle but distinct di-metal scaffold for catalytic activity. Protein Sci 29:723–743. https://doi.org/10.1002/PRO.3804
Park H, Brothers EN, Merz KM (2005) Hybrid QM/MM and DFT investigations of the catalytic mechanism and inhibition of the dinuclear zinc metallo-β-lactamase CcrA from Bacteroides fragilis. J Am Chem Soc 127:4232–4241. https://doi.org/10.1021/ja042607b
Crowder MW, Spencer J, Vila AJ (2006) Metallo-β-lactamases: novel weaponry for antibiotic resistance in bacteria. Acc Chem Res 39:721–728. https://doi.org/10.1021/ar0400241
Farnsworth,’ Olayiwola Akerele NR, Bingel AS, Soejarto DD, Zhengang & (1985) Medicinal plants in therapy*
Fransworth N (1988) Screening plants for new medicines. In: Biodiversity. pp 83–97
Sandhya B, Thomas S, Isabel W, Shenbagarathai R (2006) Ethnomedical plants used by the Valaiyan community of Piranmalai Hills (reserved forest), Tamilnadu, India - A pilot study. African J Tradit Complement Altern Med 3:101–114. https://doi.org/10.4314/ajtcam.v3i1.31145
Mohanraj K, Karthikeyan BS, Vivek-Ananth RP, et al (2018) IMPPAT: a curated database of Indian Medicinal Plants, Phytochemistry and Therapeutics. Sci Rep 8:. https://doi.org/10.1038/s41598-018-22631-z
Berman HM, Battistuz T, Bhat TN et al (2002) The Protein Data Bank. Acta Crystallogr Sect D Biol Crystallogr 58:899–907. https://doi.org/10.1107/S0907444902003451
Mohanraj K, Karthikeyan BS, Vivek-Ananth RP et al (2018) IMPPAT: a curated database of Indian Medicinal Plants. Phytochemistry and Therapeutics Sci Rep 8:4329. https://doi.org/10.1038/s41598-018-22631-z
Trott O, Olson AJ (2009) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem NA-NA. https://doi.org/10.1002/JCC.21334
O’Boyle NM, Banck M, James CA, et al (2011) Open babel: an open chemical toolbox. J Cheminform 3:. https://doi.org/10.1186/1758-2946-3-33
Adasme MF, Linnemann KL, Bolz SN et al (2021) PLIP 2021: expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res 49:W530–W534. https://doi.org/10.1093/nar/gkab294
Jespers W, Åqvist J, Gutiérrez-de-Terán H (2021) Free energy calculations for protein–ligand binding prediction. Methods Mol Biol 2266:203–226. https://doi.org/10.1007/978-1-0716-1209-5_12
Homeyer N, informatics HG-M, (2012) undefined (2012) Free energy calculations by the molecular mechanics Poisson− Boltzmann surface area method. Wiley Online Libr 31:114–122. https://doi.org/10.1002/minf.201100135
Samonis G, Karageorgopoulos DE, Maraki S et al (2012) Stenotrophomonas maltophilia infections in a general hospital: patient characteristics, antimicrobial susceptibility, and treatment outcome. PLoS ONE 7:e37375. https://doi.org/10.1371/journal.pone.0037375
Toleman MA, Bennett PM, Bennett DMC et al (2007) Global emergence of trimethoprim/sulfamethoxazole resistance in Stenotrophomonas maltophilia mediated by acquisition of sul genes. Emerg Infect Dis 13:559–565. https://doi.org/10.3201/eid1304.061378
Nagasundaram N, George Priya Doss C (2013) Predicting the impact of single-nucleotide polymorphisms in CDK2-flavopiridol complex by molecular dynamics analysis. Cell Biochem Biophys 66:681–695. https://doi.org/10.1007/s12013-012-9512-5
Dasgupta J, Sen U, Dattagupta JK (2003) In silico mutations and molecular dynamics studies on a winged bean chymotrypsin inhibitor protein. Protein Eng 16:489–496. https://doi.org/10.1093/PROTEIN/GZG070
Priya Doss CG, Chakraborty C, Chen L, Zhu H (2014) Integrating in silico prediction methods, molecular docking, and molecular dynamics simulation to predict the impact of ALK missense mutations in structural perspective. Biomed Res Int 2014: https://doi.org/10.1155/2014/895831
McCammon JA, Gelin BR, Karplus M (1977) Dynamics of folded proteins. Nature 267:585–590. https://doi.org/10.1038/267585a0
George Priya Doss C, Rajith B, Chakraboty C et al (2014) In silico profiling and structural insights of missense mutations in RET protein kinase domain by molecular dynamics and docking approach. Mol Biosyst 10:421–436. https://doi.org/10.1039/c3mb70427k
Huang M, He JX, Hu HX et al (2020) Withanolides from the genus Physalis: a review on their phytochemical and pharmacological aspects. J Pharm Pharmacol 72:649–669. https://doi.org/10.1111/JPHP.13209
Kuboyama T, Tohda C, Bulletin KKP, 2014 undefined (2014) Effects of Ashwagandha (roots of Withania somnifera) on neurodegenerative diseases. jstage.jst.go.jp 892:892–897
Dhawan M, Parmar M, Sharun K, et al (2021) Medicinal and therapeutic potential of withanolides from Withania somnifera against COVID-19. japsonline.com 11:6–013. https://doi.org/10.7324/JAPS.2021.110402
Amezian D (2018) Effect of withanolide-containing diet on gut microbial communities and AMP expression in two closely related Lepidoptera species: heliothis subflexa and
Parida PK, Paul D, Chakravorty D (2020) The natural way forward: molecular dynamics simulation analysis of phytochemicals from Indian medicinal plants as potential inhibitors of SARS-CoV-2 targets. Phyther Res 34:3420–3433. https://doi.org/10.1002/PTR.6868
Author information
Authors and Affiliations
Contributions
Shobana Sugumar: Research idea, Manuscript; Sreenithya K H: Experimental works, Result Interpretation, Manuscript; Dhananjay Jade: Result Interpretation; and Michael A. Harrison: Pictures, Manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
H, S.K., Jade, D., Harrison, M.A. et al. Identification of natural inhibitor against L1 β-lactamase present in Stenotrophomonas maltophilia. J Mol Model 28, 342 (2022). https://doi.org/10.1007/s00894-022-05336-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05336-z