
Abstract
The domain of application of the G3(MP2)//B3-SBK theory was expanded, and its efficiency was evaluated to determinate enthalpies of formation of forty-one iodine compounds. The results were compared to those obtained with the G2 theory for the same set of molecules. The G3(MP2)//B3-SBK theory showed a mean deviation and deviation standard equal to 3.7 kcal mol−1 and 6.0 kcal mol−1, respectively. The G2 theory (mean deviation = 3.1 kcal mol−1 and standard deviation = 4.9 kcal mol−1) presented a lower error and standard deviation, but at a significantly higher computational cost. For a more complete evaluation, as a secondary part of the work, it also used different functionals B3LYP, M06-2X, WB97XD, and MP2 method with four different basis sets 6-311G(d,p), LANL2DZ, jorge-ADZP, and CEP-31G(d). The best density functional/basis set combination was obtained with M06-2X/CEP-31G(d) among the three mentioned functionals. However, the produced mean deviation is significant and equal to 17.3 kcal mol−1, with a standard deviation equal to 23.0 kcal mol−1. The 6-311G(d,p) basis achieved the best performance with the MP2 method, generating an equally significant mean deviation of 12.8 kcal mol−1 with a standard deviation equal to 18.7 kcal mol−1.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Iodine is an element that belongs to group 17 of the periodic table [1]. Its compounds can be widely applied [2, 3], with emphasis on the medical-pharmaceutical area: antiseptic/disinfectant (iodine tincture, polyvinylpyrrolidone); expectorant (potassium iodide solution, KI). Potassium iodate (KIO3) is prescribed to people affected by radiation to prevent or reduce the radioactive iodine absorption by the thyroid [2]. Another great application is in the analytical area, where iodine compounds are used to determine the oils and fats unsaturation level, as one of the reagents of the Karl Fischer method or in qualitative and quantitative analyses and organic syntheses [3]. In addition, iodine has also been used in the development of new materials, such as polarizing film for liquid–crystal display (LCD) and electrolytes for dye-sensitized solar panels [1].
However, theoretical studies involving high-level calculations to obtain the thermochemical properties of iodine compounds have been little explored mainly due to computational difficulties, even though there are reliable experimental data on their compounds in the gas phase.
Recently, Leal and Custodio formulated the G3(MP2)//B3-SBK theory [4], combining the CEP pseudopotential and the respective 31G basis set developed by Stevens, Basch, and Krauss to replace the P31G basis functions used in the G3(MP2)//B3-CEP method. The main differences are in the fact that the G3(MP2)//B3-CEP theory adapts the pseudopotential to the basis set used in the corresponding original method that considers all electrons (CEP-P31G, it was the notation given to the basis function adapted in reference to Pople basis functions) and the G3(MP2)//B3-SBK theory uses the basis set developed for the respective pseudopotential (SBK or CEP-31G). Furthermore, each method has its values for the reoptimized parameters of the s, p, and sp functions in the large basis set of carbon, nitrogen, oxygen, fluorine, phosphorus, and chlorine atoms, as well as for the parameters used in the calculation of the high-level empirical correction. The G3(MP2)//B3-SBK theory was initially tested, according to the respective corresponding versions G3(MP2)//B3 [5] and G3(MP2)//B3-CEP [6], for a set of 446 systems containing representative elements up to the 4th period [H-Kr] of the periodic table. The best combination of adjusting the G3(MP2)//B3-SBK theory produced a mean absolute error for all tested properties of 1.43 kcal mol−1. This deviation is essentially equal to that presented by the all-electron theory G3(MP2)//B3 (1.41 kcal mol−1) with savings of up to 45% CPU time and it has better accuracy than the corresponding theory that uses adapted pseudopotential G3(MP2)//B3-CEP (1.60 kcal mol−1) with equivalent CPU time. Unlike the G3(MP2)//B3 and G3(MP2)//B3-CEP theories, the G3(MP2)//B3-SBK theory allows the calculation of thermochemical properties of compounds for some elements of the 5th line of the periodic table, like Sn and Sb. Thus, the idea of expanding the application domain of this method for calculations of compounds containing heavier elements, such as iodine, arose due to its relevance in many chemical and biological processes.
Other papers can be used as a reference to demonstrate the efficiency of using composite methods. As an example, Silva [7, 8] recently used the composite methods EnAt1, EnAt2, G3X-CEP, G3X(CCSD)-CEP, and G4 [9] to determine the enthalpies of formation of 29 aluminum compounds. The Stuttgart/Dresden pseudopotential was used in the G3(MP2,CSSD,rel) theory for 20 compounds containing transition elements from scandium (Sc) to zinc (Zn) [10]. The G4(CEP) theory [11, 12] was used to accurately determine the pKa of 22 monocarboxylic acids. Other examples of applications can be found in the study of Diels–Alder reaction mechanisms [13], phenol nitration [14], rotation barriers [15], and among others.
The objective of this research was to expand the application domain of the G3(MP2)//B3-SBK theory [4] to calculate enthalpies of formation of iodine-containing compounds. The calculations efficiency was performed comparatively to accurate experimental data and calculated with the G2 theory [16, 17]. Other conventional methods (e.g., DFT, MPn) were combined with different basis sets (e.g., Pople, Dunning) for a more global assessment of errors produced by conventional calculations regarding experimental data available in the literature.
Computational methods
All calculations were performed using the Gaussian 09 software [18]. The enthalpies of formation were calculated, at 298.15 K and 1 atm, according to the standard procedure established in the literature [19]: (a) The first step is to calculate the enthalpies of formation \(({\Delta H}_{f}^{0} \left(M,0 K\right)\)) of the molecule \(\left(M\right)\) at 0 K from the molecular atomization energy \(\left(\sum {D}_{0}(M)\right)\) and heats of formation of the atoms \((\sum_{atoms}x{\Delta H}_{f}^{0} \left(X, 0 K\right))\) at 0 K:
where \(\sum_{atoms}x{\varepsilon }_{0} \left(X\right)\) is the sum of the energies of the constituent atoms, \({\varepsilon }_{0} \left(M\right)\) is the total energy, and \({\varepsilon }_{ZPE}(M)\) is the zero-point energy of the molecule. \(X\) is each element which makes up M, and \(x\) is the number of atoms of \(X\) in the molecule \(M\). (b) The second step is to calculate the enthalpies of formation \(({\Delta H}_{f}^{0} \left(M, 298 K\right))\) at 298 K:
where \({(H}_{M}^{0} \left(298 K\right)-{H}_{M}^{0} \left(0 K\right))\) is the molecular enthalpy correction and \(\sum_{atoms}x({H}_{X}^{0}\left(298 K\right)- {H}_{X}^{0} \left(0 K\right))\) is the sum of the enthalpy corrections of the atomic elements. Table 1 shows values used for enthalpies of formation of the elements at 0 K and respective thermal corrections (\({\mathrm{H}}^{0} (298\mathrm{ K})-{\mathrm{H}}^{0} (0\mathrm{ K})\)), at 298.15 K (25 °C) and 0.1 MPa (1 bar), both were taken from references [8, 19,20,21].
Knowing that the SBK or CEP–31G basis functions [22, 23] were not developed with polarization functions, similar to the procedure adopted by Leal and Custodio to include tin (Sn) and antimony (Sb) atoms [4], the adjustment of the polarization function for iodine was performed while minimizing the energies of a subset of 14 molecules through the optimization at B3LYP/CEP–31G(d) level. For this adjustment, systems available at WebNIST [24] were selected which presented experimental values of standard enthalpy of formation in the gas phase with an uncertainty of the order of ± 1 kcal mol−1: HI, CH3I, C2H5I, CIN, CH3IS, C3H5I, C2H5IS, C4H9I, C2H4ICl, C2H3IO, C4H7IO, C7H5IO, CF3I, and C2H2F3I. The obtained value of 0.270 for the polarization function exponent of the iodine atom is similar to the corresponding values of 0.266 (G2 theory, all-electron) [17], 0.279 (ECP(HW), the shape-consistent orbital-adjusted ECPs of Hay and Wadt) [17, 25], 0.267 (ECP(S), energy-adjusted ECPs of the Stuttgart group (S)) [17, 26] that are the values of energy-optimized exponents of polarization d functions for augmentation of the valence basis sets in all-electron (AE) and effective core potential (ECP) calculations iodine-containing species. The values of the polarization functions’ exponents, used together with the CEP-31G set, for the other elements were taken from the Leal and Custodio work [4].
The energy G3(MP2)//B3–SBK [4] is determined through the equation:
in which, E[QCISD(T)/CEP-31G(d)] is the reference energy; ΔEG3MP2large = E[MP2/CEP–G3MP2large] – E[MP2/CEP–31G(d)] is the energy component calculated with the basis function CEP-G3MP2large, included to correct the effects of extending the basis sets; ESO is the spin–orbit correction for atoms whose values were extracted from the literature [9, 17]; EHLC is an empirical correction represented by EHLC = − Anβ − B(nα − nβ) for molecules (being A = 8.849 and B = 4.495, mHartree) and EHLC = − Cnβ − D(nα − nβ) for atoms and atomic ions (being C = 9.436 and D = 1.586, mHartree), in which nα and nβ are the number of valence electrons with alpha and beta spins, respectively; Etherm is the energy component that contains the zero point correction and thermal effects related to translational, rotational, and vibrational motions. All vibrational frequencies were scaled by a factor of 0.96. The cc-pV5z-PP basis set [27] was considered the CEP-G3MP2large for the iodine atom.
The energy G2 [16, 17] is obtained through an improvement of the G1 energy [28] stated by:
in which, E0(G1) is the energy obtained through the G1 theory; Δ = E[MP2/6–311 + G(3df,2p)] – E[MP2/6-311G(2df,p)] – E[MP2/6–311 + G(d,p) + E[MP2/6-311G(d,p)] and npair is the number of valence electron pairs. The detailed step-by-step to obtain energy E0(G1) can be directly verified in Pople et al. [28]. To obtain the G2 energy of compounds involving the iodine element, the protocol established by Glukhovtsev et al. [17] was followed.
In addition to the G3(MP2)//B3–SBK and G2 composite methods, the performance of three popular functionals B3LYP [29, 30], M06-2X [31], WB97XD [32], and the MP2 method [33] with the basis sets 6-311G(d,p) [17, 34, 35], LANL2DZ [25, 36], jorge-ADZP [37, 38], and CEP-31G(d) were also evaluated as formulated in this research, since these methodologies also allow direct application to systems containing iodine, which contributes to a global assessment.
Results and discussion
Table 2 shows the errors set (ΔH0f (exp) – ΔH0f (calc)) calculated concerning experimental values of standard enthalpy of formation (high precision in the gas phase) available in WebNIST [24] for 41 iodine compounds with the G2 and G3(MP2)//B3-SBK theories. Besides, mean absolute deviation (MAD) and standard deviation (SD) values are also reported.
The G3(MP2)//B3-SBK method presented a mean absolute deviation equal to 3.7 kcal mol−1 and a standard deviation equal to 6.0 kcal mol−1, while the G2 method presented values of 3.1 kcal mol−1 and 4.9 kcal mol−1 respectively. The deviations obtained with the G3(MP2)//B3-SBK theory are greater when compared to those obtained with the G2 theory, but achieving the G3(MP2)//B3-SBK energy involves fewer steps in addition to the smallest computational cost. There is a balance in the individual performance between these two composite methods, and there are 17 systems that present errors (regarding the experimental data) outside the range of ± 2 kcal mol−1 when obtained with the G2 theory, against 19 systems when calculated with the G3(MP2)//B3-SBK theory.
According to the literature [4, 6, 42], obtaining thermochemical or spectroscopic properties of compounds containing fluorine atoms usually exhibits unusual behavior and it is still a challenge to predict these properties with high accuracy, even with the use of composite methods. Even the G3(MP2)//B3-SBK method with the scaling of experimental atomization energies still showed high deviations for enthalpies of formation C2F4 (4.6 kcal mol−1), PF3 (− 7.0 kcal mol−1), SF6 (− 8.7 kcal mol−1), ClFO3 (− 18.7 kcal mol−1), and ionization energies B2F4 (8.7 kcal mol−1), BF3 (− 5.2 kcal mol−1), and CH3F (− 5.0 kcal mol−1) of fluorinated molecules. In this work, high deviations respectively with the G2 and G3(MP2)//B3-SBK theories were also observed for the C2H3F2I (5.6 and 4.3 kcal mol−1), C2H2F3I (6.9 and 4.8 kcal mol−1), CF3I (4.9 and 3.3 kcal mol−1), F5I (5.1 and 17.0 kcal mol−1), and F7I (20.8 and 25.9 kcal mol−1) systems. Due to the low uncertainty associated with experimental measurements, for example ΔH0f (exp): C2H2F3I = − 155.0 ± 0.9 kcal mol−1, ΔH0f (exp): CF3I = − 140.8 ± 0.1 kcal mol−1 [39, 40], ΔH0f (exp) F5I = − 200.8 ± 0.4 kcal mol−1 [41], ΔH0f (exp): F7I = − 229.7 ± 0.5 kcal mol−1 [41], higher level calculations associated with the use of more extended basis sets are welcome in an attempt to better reproduce the experimental behavior of these molecules. However, the high computational cost often limits the use of even more rigorous composite methods. In addition, more than one experimental information can often be found for the same system, making it difficult to choose a reference standard (for example: most recent measurement, measurement with lower uncertainty, measurement leading to a lower calculated error, among others). In this work, the measure associated with the lowest experimental uncertainty was chosen as the reference standard.
Recently, from a modification of the G4MP2 theory [43], E. Sookhaki and M. Namazian developed the G4MP2-ECP method [44] specifically to allow calculations involving the iodine atom. Among the various tested properties, they evaluated the performance of the G4MP2-ECP method for standard enthalpy of formation of a set of 19 iodinated organic compounds. Table 3 shows the performance of G2 and G3(MP2)//B3-SBK theories compared to experimental data and the G4MP2-ECP method. The best performance was achieved with the G2 theory (MAD = 0.8 kcal mol−1 and SD = 1.1 kcal mol−1), followed by the G3(MP2)//B3-SBK theory (MAD = 1.0 kcal mol−1 and SD = 1.4 kcal mol−1) and finally G4MP2-ECP (MAD = 1.6 kcal mol−1 and SD = 2.0 kcal mol−1).
Regarding the functional/basis set combination (Appendices Tables 4 and 5), the smallest deviations were achieved by combining the different functionals to the CEP-31G(d) basis set, with M06-2X (MAD = 17.3 kcal mol−1 and SD = 23.0 kcal mol−1) as the functional with the best performance against the experimental results, followed by WB97XD (MAD = 20.1 kcal mol−1 and SD = 28.0 kcal mol−1) and B3LYP (MAD = 24.5 kcal mol−1 and SD = 29.9 kcal mol−1). Functional M06-2X stands out above all for the low deviations achieved in fluorinated systems such as C6F5I (7.8 kcal mol−1), F5I (− 6.4 kcal mol−1), F7I (− 20.8 kcal mol−1), and FI (0.7 kcal mol−1) compared to B3LYP and WB97XD functionals. It is also noteworthy that the M06-2X and WB97XD functionals combined with the CEP-31G(d) set produced the smallest deviation (only, − 5.1 kcal mol−1), among all the tested methodologies, for the C2H3IN2O4 system. This evaluation is particularly useful as it can direct and drive the development of an alternative composite method using the M06-2X functional combined with the CEP-31G(d) set.
Regarding the combination of the MP2 method (Appendix Table 5), with the four tested basis sets, the smallest deviations (MAD = 12.9 kcal mol−1 and SD = 18.7 kcal mol−1) were achieved when the 6-311G(d,p) set was used. This is mainly due to low errors, including for (INO), (CIN3O6), and (C2H3IN2O4) systems that showed a divergence of only − 6.8; 4.8 and 9.0 kcal mol−1 concerning the experimental data, respectively. However, systems like (F5I) e (F7I) still showed high deviations regarding the experimental data, − 50.5 and − 57.0 kcal mol−1 respectively. The second smallest deviation (MAD = 21.0 kcal mol−1 and SD = 25.5 kcal mol−1) was achieved by combining MP2/jorge-ADZP, followed by MP2/CEP-31G(d) (MAD = 25.7 kcal mol−1 and SD = 29.8 kcal mol−1) and, finally, MP2/LANL2DZ (MAD = 53.6 kcal mol−1 and SD = 63.8 kcal mol−1).
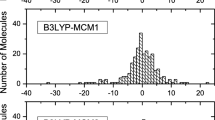
Figure 1 shows the mean absolute deviation (MAD) for all methodologies tested considering the set of enthalpies of formation of the forty-one iodine compounds studied. Appendix Table 6 summarizes the energy of atoms for each methodology evaluated in this research.

Mean absolute deviation (MAD) for all methodologies tested considering the set of enthalpies of formation of the forty-one iodine compounds studied: a B3LYP, b M06-2X, c WB97XD, and d MP2
Conclusion
In this paper, the domain of application of the G3(MP2)//B3-SBK theory was extended to study the enthalpies of formation of 41 iodine compounds. For the first time for this method and for the iodine element, was added a polarization function in the CEP–31G basis set, and adjusting its exponent, the value of 0.270 was obtained, which is in good agreement with other corresponding values found in the literature. The G3(MP2)//B3-SBK method showed a mean absolute deviation equal to 3.7 kcal mol−1 and a standard deviation equal to 6.0 kcal mol−1, while the G2 method presented values of 3.1 kcal mol−1 and 4.9 kcal mol−1 respectively. The deviations obtained with the G3(MP2)//B3-SBK theory are greater when compared to those obtained with the G2 theory, but achieving the G3(MP2)//B3-SBK energy involves fewer steps in addition to the smaller computational cost. There is a balance in the individual performance between these two composite methods, there are 17 systems that present errors (regarding the experimental data) outside the range of ± 2 kcal mol−1 when obtained with the G2 theory, against 19 systems when calculated with the G3(MP2)//B3-SBK theory.
Regarding the standard enthalpy of formation of the nineteen iodized organic compounds set, previously studied by E. Sookhaki and M. Namazian with the G4MP2-ECP method (MAD = 1.6 kcal mol−1 and SD = 2.0 kcal mol−1), it was found that the G2 (MAD = 0.8 kcal mol−1 and SD = 1.1 kcal mol−1) and G3(MP2)//B3-SBK (MAD = 1.0 kcal mol−1 and SD = 1.4 kcal mol−1) theories showed smallest deviations with respect to the experimental data.
Concerning the performed additional tests combining different functionals B3LYP, M06-2X, WB97XD, and the MP2 method with different basis sets 6-311G(d,p), LANL2DZ, jorge-ADZP, and CEP-31G(d), the best combination functional/basis set was obtained with M06-2X/CEP-31G(d) producing a MAD = 17.3 kcal mol−1 and SD = 23.0 kcal mol−1. The best performance with the MP2 method was achieved with Pople 6-311G(d,p) basis generating a MAD = 12.9 kcal mol−1 and SD = 18.7 kcal mol−1. These additional tests mainly serve to reaffirm the efficiency of the composite methods in the accurate determination of thermochemical properties, including now the case of the standard enthalpy of formation of iodine compounds.
References
Tatsuo K (2015) Iodine chemistry and applications. Wiley, Hoboken
Santos VM, Afonso JC (2013) QNEsc 35:297–298
Santos VM, Afonso JC (2012) Quim Nova 35:398–402
Leal RC, Custodio R (2019) Comput Theor Chem 1149:1–7
Baboul AG, Curtiss LA, Redfern PC, Raghavachari K (1999) J Chem Phys 110:7650–7657
Rocha CMR, Pereira DH, Morgon NH, Custodio R (2013) J Chem Phys 139:184108–184119
Silva CS (2020) Theor Chem Acc 139:135–142
Silva CS, Custodio R (2018) Theor Chem Acc 137:24–32
Curtiss LA, Redfern PC, Raghavachari K (2007) J Chem Phys 126:084108–084119
Silva CS, Custodio R (2015) Rev Proc Q 9:66–67
Silva CS, Custodio R (2019) J Phys Chem A 123:8314–8320
Silva CS, Pereira DH, Custodio R (2016) J Chem Phys 144:204118–204126
Leal RC, Pereira DH, Custodio R (2018) Comput Theor Chem 1123:161–168
Rocha CMR, Rodrigues JAR, Moran PJS, Custodio R (2014) J Mol Model 20:2524–2531
Filho SQA, Costa AMF, Ribeiro IHS, Custodio R, Pereira DH (2019) Comput Theor Chem 1166:112589–112595
Curtiss LA, Raghavachari K, Trucks GW, Pople JA (1991) J Chem Phys 94:7221–7230
Glukhovtsev MN, Pross A, McGrath MP, Radom L (1995) J Chem Phys 103:1878–1885
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09 (Revision A.02)
Curtiss LA, Raghavachari K, Redfern PC, Pople JA (1997) J Chem Phys 106:1063–1079
Luo YR (2007) Comprehensive handbook of chemical bond energies. CRC Press, Taylor and Francis Group, Boca Raton
Wagman DD, Evans WH, Parker VB, Schumm RH, Halow I, Bailey SM, Churney KL, Nuttall RL (1982) The NBS tables of chemical thermodynamic properties selected values for inorganic and C1 C2 organic substance in SI units. J Phys Chem Ref Data 11:1–392
Stevens WJ, Basch H, Krauss M (1984) J Chem Phys 81:6026–6033
Stevens WJ, Krauss M, Basch H, Jasien PG (1992) Can J Chem 70:612–630
Linstrom PJ, Mallard WG (2022) NIST CHEMISTRY WebBook, NIST Standard Reference Database Number 69
Wadt WR, Hay PJ (1985) J Chem Phys 82:284–298
Bergner A, Dolg M, Küchle W, Stoll H, Preuss H (1993) Mol Phys 80:1431–1441
Peterson KA, Figgen D, Goll E, Stoll H, Dolg M (2003) J Chem Phys 119:11113–11123
Pople JA, Head-Gordon M, Fox DJ, Raghavachari K, Curtiss LA (1989) J Chem Phys 90:5622–5629
Becke AD (1988) Phys Rev A 38:3098–3100
Lee C, Yang WT, Parr RG (1988) Phys Rev B 37:785–789
Zhao Y, Truhlar DG (2008) Theor Chem Acc 120:215–241
Chai JD, Head-Gorgon M (2008) Phys Chem Chem Phys 10:6615–6620
Møller C, Plesset MS (1934) Phys Rev 46:618–622
McLean AD, Chandler GS (1980) J Chem Phys 72:5639–5648
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) J Chem Phys 72:650–654
Dunning TH Jr, Hay PJ (1977) Gaussian basis sets for molecular calculations. In: Schaefer HF (ed) Methods of Electronic Structure Theory. Modern Theoretical Chemistry, Springer Boston, Massachusetts, pp 1–27
Neto AC, Muniz EP, Centoducatte R, Jorge FE (2005) J Mol Struc 718:219–224
Oliveira PJP, Barros CL, Jorge FE, Neto AC, Campos M (2010) J Mol Struc 948:43–46
Ruscic B, Pinzon RE, Morton ML, Laszewski GV, Bittner S, Nijsure SG, Amin KA, Minkoff M, Wagner AF (2004) J Phys Chem A 108:9979–9997
Ruscic B, Pinzon RE, Laszewski GV, Kodeboyina D, Burcat A, Leahy D, Montoya D, Wagner AF (2005) J Phys Conf Ser 16:561–570
Settle JL, Jeffes JHE, O’Hare PAG, Hubbard WN (1976) J Inorg Nucl Chem 28:135–140
Curtiss LA, Raghavachari K, Redfern PC, Pople JA (2000) J Chem Phys 112:7374–7383
Curtiss LA, Redfern PC, Raghavachari K (2007) J Chem Phys 127:124105–124112
Sookhaki E, Namazian M (2021) J Mol Graphics Modell 108:107985–107991
Richard L, Gaona X (2011) Geochem Cosmochim Acta 75:7304–7310
Bodi A, Shuman NS, Baer T (2009) Phys Chem Chem Phys 11:11013–11021
Lago AF, Kercher JP, BÖdi A, Sztáray B, Miller B, Wurzelmann D, Baer T (2005) J Phys Chem 109:1802–1809
Dávalos JZ, Notario R, Cuevas CA, Oliva JM, Saiz-Lopez A (2017) Comput Theor Chem 1099:36–44
Zherikova KV, Verevkin SP (2019) J Therm Anal Calorim 138:4045–4059
Acknowledgements
The authors wish to thank Dr. Rogério Custodio for the help in the discussion and suggestions. Ysa Beatriz Dantas Marinho and Maria Andreizi Monteiro de Andrade thank the Institutional Research Support Program and the Research and Innovation Dean (PROPI) from IFRN for the research grants granted in the Research Notices nº 01/2019 (1st Call)—PROPI/RE/IFRN—Development of Research and Innovation, Projects nº 04/2020—PROPI/RE/IFRN—Research and Innovation Projects with Support, respectively. The authors would like to acknowledge the National Center of High-Performance Computing in Ceará (CENAPAD-UFC) for access to their computational facilities.
Author information
Authors and Affiliations
Contributions
Leal, R. C. and Sousa, I. L. analysis of data and writing. Marinho, Y. B. D. and Andrade, M. A. M. performed the calculations.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper belongs to Topical Collection XXI − Brazilian Symposium of Theoretical Chemistry (SBQT2021)
Appendix
Appendix
Table 4
Table 5
Table 6
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Leal, R.C., Marinho, Y.B.D., de Andrade, M.A.M. et al. Determination of the standard enthalpy of formation of iodine compounds through the G2 and G3(MP2)//B3-SBK theories. J Mol Model 28, 246 (2022). https://doi.org/10.1007/s00894-022-05243-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05243-3