
Abstract
In this study, 32 energetic compounds were designed using oxadiazoles (1,2,5-oxadiazole, 1,3,4-oxadiazole) as the parent by inserting different groups as well as changing the bridge between the parent. These compounds had high density and excellent detonation properties. The electrostatic potentials of the designed compounds were analyzed using density functional theory (DFT). The structure, heat of formation (HOF), density, detonation performances (detonation pressure P, detonation velocity D, detonation heat Q), and thermal stability of each compound were systematically studied based on molecular dynamics. The results showed that the -N3 group has the greatest improvement in HOF. For the detonation performances, the directly linked -N=N- and -NH-NH- were beneficial when used as a bridge between 1,2,5-oxadiazole and 1,3,4-oxadiazole, and it can also be found that bridge changing had little effect on the trend of detonation performance, while energetic groups changing influenced differently. In general, the introduction of nitro groups contributes to the improvement of the detonation performance of the compounds. In this study, the compounds containing the highest amount of nitro groups were found to have better detonation performance than their counterparts and were not significantly different from RDX and HMX.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, the design and synthesis of new high energy density materials is an important part of research in the field of energetic materials [1,2,3,4]. High energy density materials (HEDMs) are compounds of a given volume that can generate a considerable amount of energy simply through the creation and breaking of chemical bonds within the molecule. High energy density materials have a series of advantages such as high energy density, intensity, and stability. In addition, to complete the research on HEDMs, high nitrogen energetic materials as a new type of energetic material have also received more and more attention due to their considerable positive heat of formation, high density, excellent detonation properties (detonation velocity and detonation pressure), and admissible thermal stability [5].
1,2,5-oxadiazole (furazan), with the molecular formula C2H2ON2, is a five-membered ring. Zelinskii [6,7,8] Institute of Organic Chemistry, Russian Federation Academy of Science had synthesized a variety of furazan energetic compounds after more than 20 years of research. Chaoyang Zhang [9] reported nitro-furazan to increase in density of 0.06–0.08 g cm3 and detonation velocity of about 300 m s−1. 1,3,4-oxadiazole is an isomer of furazan with a low enthalpy of production; however, the synthesis of energetic compounds with high nitrogen content using 1,3,4-oxadiazole can reduce the sensitivity of energetic compounds and improve the oxygen balance. Shreeve et al. reported the use of 1,3,4-oxadiazole to stabilize the nitrofurazone ring [10]. Based on 1,3,4-oxadiazole and the nitrofurazone backbone, a series of high nitrogen energetic salts were synthesized with good detonation properties and satisfactory susceptibility and stability (detonation velocities >7493 m s−1, detonation pressures >20.4 GPa, IS = 15 J, FS = 120 N). Lu Ming et al. concluded that the compounds formed by incorporating azo 1,3,4-oxadiazole with nitramino furazan and nitro-furazan have significant thermal stability (Td > 233 °C, Td(RDX) = 204 °C), and 1,3,4-oxadiazole is expected to contribute to the construction of thermally stable oxadiazole molecules [11]. Qian Wang combined isomers of oxadiazoles into bicyclic compounds using a simple synthetic strategy and obtained energetic materials with excellent detonation properties. (3,3′-dinitramino-4,4′-bifurazane features a detonation velocity of 9086 m s−1; 3′-dinitroxazafurazan, which includes two nitro-furazan fragments, has an excellent detonation performance, D = 9390 m s−1; P = 40.5 GPa.)
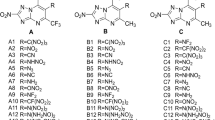
In this work, we conducted systematic research on 1,3,4-oxadiazole and 1,2,5-oxadiazole as a backbone with different bridges and various energetic groups inserted (shown in Scheme 1). Density functional theory (DFT) and electrostatic potential (ESP) studies were used to analyze these compounds, along with their HOFs, densities, and detonation properties.

Designed energetic molecules based on oxadiazoles
Computational methods
In this work, all quantum mechanical calculations were done using Gaussian 16 software [12]. Structural optimization of the designed compounds was at B3LYP/6-311G (d,p) level [13,14,15], with all structures reaching “no imaginary frequency” to achieve stable structures. The frontier molecular orbitals (FMOs) and the electrostatic potential were both calculated at the identical theoretical level. After that, the total energy of the molecules was determined at the B3LYP/def2-TZVPP level. The reactions and associated schemes designed to anticipate the gas-phase HOFs of the designed compounds were as follows (shown in Scheme 2):

Representative isodesmic reactions for designed compounds
The advantage of designing isodesmic reaction calculations was that the types and numbers of various bonds of the reactants and products in the isodesmic reaction are correspondingly the same, which can effectively reduce errors in the calculation of HOFs. HOFs at a certain temperature (∆H298K) was calculated by the following equation:
For the ∆H298K, HOFs in Eqs. (1) and (2) were calculated. ∆Hf,P and ∆Hf,R are the HOFs of the reactants and products, respectively. ΔE0 is the single-point energy, changes in energy from product to reactant; ΔZPE is the difference between the zero-point energy (ZPE) of the target product and reactant; ΔHT is thermal correction starting with 0 will 298 K; n is energy group in numbers; ∆(PV) equals ∆nRT.
Most of the energetic compounds are not in the gas state but the solid state. In the calculation of detonation performance (heat of detonation, detonation velocity, detonation pressure), the calculation of the solid-phase HOFs (ΔHf,solid) was the first step. According to Hess’s law, the relationship between ΔHf,solid and ΔHf,gas can be expressed by the following formula [16]:
The relational equation proposed by Politzer et al. can be used to calculate the ΔHsub [17, 18]:
where a, b, and c are constants of 0.000267 kcal mol−1 Å−4, 1.650 kcal mol−1, and 2.966 kcal mol−1, respectively. A denotes the surface area of a molecule with an electron density of 0.001e Bohr−3 equivalents. ν is a measure of the balance between positive and negative regions on the surface, \( {\sigma}_{\mathrm{tot}}^2 \) represents a measurement of the electrostatic potential variability of the molecular surface, which can be obtained by Multiwfn [19].
The computational formula proposed by Politzer et al. [20]. can be used to calculate the density of the detonation velocity and pressure.
α, β, and γ are constants with values of 0.9183, 0.0028, and 0.0443, correspondingly. M represents the molar mass of the molecule (g mol−1) and V represents the volume of the molecule (m3 mol−1). ν \( {\sigma}_{\mathrm{tot}}^2 \) indicate the same as above.
Detonation performance (detonation velocity and detonation pressure) is calculated according to the Kamlet-Jacobs equation [21].
Here, D stands for blast velocity (km s−1), and the P indicates the burst pressure (GPa). N, \( \overline{M} \), and Q are the number of moles of initiating gases per gram of explosive (mol g−1), the average combined molecular weight of these gases (g mol−1), and the heat of detonation (cal g−1), respectively.
Results and discussion
Electronic structures
The frontier molecular orbital, i.e., the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO), can gather suitable information on optical polarizability, dynamic stability, and reactivity [22,23,24,25]. The HOMO, LUMO, and their energy gaps (ΔELUMO–HOMO) of the designed structures are shown in Table 1.
From Table 1, it is not difficult to find that for HOMO, when inserting different energetic groups, the four series of A, B, C, and D have similar trends in energy level changes. Particularly for each series, HOMO energy increases with the substitution of -NH2 and -NHNH2, while the HOMO energy decreased when -CH(NO2)2, -C(NO2)3, and -NO2 were inserted. The ranking of the contribution of each group to HOMO is as follows: -NH2 ≈ -NHNH2 > -N3 > -ONO2 > -NHNO2 > -CH(NO2)2>-NO2>-C(NO2)3. The same is true for the LUMO energy level. Overall, the bridge has little effect on the trend of HOMO and LUMO energy changes for each designed compound. However, comparing to other bridging links, D series (-NH-NH-) has the highest LUMO energy level except for D6, which meant the backbone played a proactive role in LUMO.
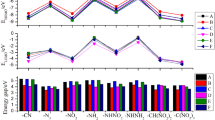
The trend of ΔELUMO–HOMO for each series can also be seen in Table 1. It is easy to find that three series A, B, and C have similar trends in ΔELUMO–HOMO, in which the gap is relatively small when the energetic groups are -N3, -NHNH2, and -ONO2, the structure is relatively less stable, and the intensity of interatomic interactions is higher. It indicates a shift towards lower frequencies in their electronic absorption spectra. The difference is that in the D series, the ΔELUMO–HOMO changes drastically, yet the D7 (4.26 eV) and B7 (4.29 eV) have similar gap values. Figure 1 shows the varied trends of the ΔELUMO–HOMO of A series (directly linked) in the designed compounds. Clearly, all compounds of A series have comparatively larger energy gaps from 4.55 to 5.38 eV, meaning that these molecules possess good chemical stabilities. The findings indicate that the parent structure is the main influence of the ΔELUMO–HOMO variation in series A, B, and C, the order of these series in average ΔELUMO–HOMO can be written as follows: directly linked >-C=C->-N=N-; the energetic group is the main influence of the D series. According to previous studies, the smaller the ΔELUMO–HOMO in a similar structure, the more unstable the structure is, which implies that the polarization rate is larger when the energy difference between occupied and non-occupied orbitals is smaller [25]. In the case where the bridges are both conjugated, the introduction of the -N=N- makes ΔELUMO–HOMO smaller than when -C=C- is introduced. From all designed compounds, D2 (5.66 eV) has the highest ΔELUMO–HOMO, while C2 (3.35 eV) has the lowest. In other words, the volatility of compound C2 is more evident than other compounds.

The variation trends of ΔELUMO–HOMO of the designed compounds
Heat of formation
The heat of formation (HOFs) is generally employed as an indicator with regard to the “energy content” of a HEDM [26]. Additionally, HOF is a key parameter that predicts the detonation properties (especially the heat of detonation) of energetic materials. In order to predict accurate HOFs, calculations are usually performed by atomization reactions (mainly for small molecules) or isodesmic reactions (mainly for complex compounds). Table 2 presents calculated total energies (E0), zero-point energies (ZPE), and thermal corrections (HT) for the reference compounds in the isodesmic reactions.
Table 3 lists the total energies, ZPEs, thermal corrections, ΔHf,gas, A, v, \( {\sigma}_{tot}^2 \), ΔHsub, and ΔHf, solid of designed materials. Except for A8 (−97.87 kJ mol−1) and B8 (−67.23 kJ mol−1), it is shown that almost all compounds have positive ΔHf,gas range from A5 (18.95 kJ mol−1) to C1 (1248.67 kJ mol−1). About ΔHf,solid, 24 compounds have positive HOFs (except for B8, −203.34 kJ mol−1; A8, −202.70 kJ mol−1; B5, −122.96 kJ mol−1; A5, −120.25 kJ mol−1; D8, −112.99 kJ mol−1; D5, −27.33 kJ mol−1; B7, −11.75 kJ mol−1; A7, −4.56 kJ mol−1) in the range from B6 (12.97 kJ mol−1) to C1 (1123.86 kJ mol−1). It is noted that for each designed series, -N3 energetic group has the most important effect in improving HOFs; however, -CH(NO2)2 and -ONO2 both play a negative role to HOFs. Overall, after comparing with solid-phase HOFs of common HEDMs (HMX 272.6 kJ mol−1), 12 compounds are higher than HMX. These high HOFs can give a great contribution to the detonation properties (heat of detonation, detonation pressure, detonation velocities).
Figure 2 shows the gas-phase heat of formation for the designed structures. It can be observed that the HOFs of each series change due to the variation of the high-energy groups. The four series of compounds, A, B, C, and D, have similar trends in HOF changes, and it can be assumed that energetic groups have a great influence on HOF values. When two groups -N3 and -NHNH2 are used as substituents, HOFs of each series are higher. The order of influences about different energetic groups is as follows: -N3 > -NHNH2 > -NHNO2 > -NH2 > -C(NO2)3 > -NO2 > -CH(NO2)2 > -ONO2. The results are similar to those of the analysis of the electronic structure above. The order of contributions to HOF by bridges is as follows: -N=N- > directly linked > -C=C- ≈ -NH-NH-. The more N-H bonds in the structure, the larger the HOF value is.

The variation trends of ΔHf,gas of the designed compounds
Detonation property
Oxygen balance (OB), density (ρ), the heat of detonation (Q), detonation velocity (D), and detonation (P) are the detonation properties and are shown in Table 4. Clearly, after calculating detonation performances of common energetic materials (TNT, RDX, HMX), the results are in agreement with the experimental data, proving that the calculation method is feasible. OB represents the degree to which all carbon atoms in the molecule are oxidized to carbon dioxide; hydrogen atoms are oxidized to water, and the increased production of CO2 and H2O represents the increased heat of formation [32, 33]. At the same time, an excessively high OB results in a large amount of energy being taken away, so the ideal value for OB should be zero. Apparently, A6 (7.3%), C6 (6.9%), D6 (3.4%), C8 (0.0%), A8 (0.0%), D8 (−5.5%), and B6 (−10.4%) own acceptable values of OB. It can infer that -ONO2 and -C(NO2)3 play an important role in dominating OB among all compounds.
The densities of all designed compounds range from 1.5 to 2.0 g cm−3. When -CH(NO2)2, -C(NO2)3, -NO2, and -ONO2 are inserted, the density of the compounds is comparable to that of TNT, RDX, etc. However, the compounds with the substituent -C(NO2)3 have the highest densities.
Detonation velocity (D) and detonation pressure (P) are two main parameters to determine detonation performance of high-energy materials [34]. Table 4 lists detonation performance of the designed structures and representative HEDMs. Four series of designed structures have good performance in detonation velocity (between 6.10 and 9.41 km s−1) After a comparison, the velocity values of RDX and HMX are found to have a close value than 5 compounds (A8, B6, D6, C6, and A6), and it can be regarded that -C(NO2)3 gives the greatest contribution to the detonation velocity. This is also consistent with the results of the OB analysis. In the four series of designed objects, it can be seen that the -C=C- bond has a small detonation velocity when joined, and the -NH-NH- and -N=N- join have a small difference in detonation velocity (except D5).
Compounds with a substitution base of -NH2 perform poorly in detonation velocity and are also relatively low in detonation pressure. The detonation pressure of the designed structure is basically in the range of 21.32–41.86 GPa, removing B4 (14.82 GPa), B2 (14.86 GPa), D2 (18.91 GPa), C2 (19.10 GPa), and A2 (19.28 GPa). Similarly, the detonation pressure is greatest at the insertion of the -C(NO2)3 energetic group. It can also be seen from Fig. 3d that the order of effect of substituents in the pressure is as follows: -C(NO2)3>-ONO2 ≈ -CH(NO2)2 ≈ -NHNO2 ≈ -NO2>-N3 >-NHNH2>-NH2. For detonation pressure, different from the similar level of three series A, C and D, the level of B is smaller particularly.

a, b, c, and d stand for variation trends of ρ, D, P, and Q of the designed compounds
When there are more bi-atomic molecular gases CO and H2 in the product, it indicates that less CO2 and H2O will be produced, which will lead to a decrease in Mave and a decrease in Q [22]. It is clear from Fig. 3b that the structure with the -C(NO2)3 or -N3 substituents in the same series has the largest Q and the smallest -NH2. When a single bond (A series) is added, the heat of detonation is relatively larger than the other bridging.
Thermal stability
There is an essential problem in the design and synthesis of new high-energy materials: whether HEDMs have sufficient dynamic stability to be of practical interest. The bond dissociation energy (BDE) can be regarded as an indispensable indicator in understanding thermal stability and decomposition process of high-energy materials [35]. Moreover, previous studies [36, 37] on the BDE of nitro compounds especially nitroaromatic and nitramine molecules showed that the BDE of energetic material is related to its sensitivity and stability directly, and the smaller the BDE is, the more sensitive the compound is. Generally speaking, the smaller the bond dissociation energy required for bond dissociating, the weaker the bond is, which is also one of the ways to find out the trigger bond. Bond order is one of the quantitative descriptions of chemical bonds, which can be used for understanding the molecular electronic structure and predicting the molecular reactivity and stability Therefore, certain relative bonds are selected as the dissociating bond to calculate BDE according to the bond overlap population Laplacian bond order (LBO) [38]. Table 5 shows the BDE of almost designed compounds.
It is interesting that as listed in Table 5, most of the trigger bonds in the B series (-C=C-) are chemical bonds between the parent and the energetic group, and the value of BDE is relatively high (range from 249.89 to 468.45 kJ mol−1); thus, series B is more stable. The analysis also shows that most of the trigger bonds in series D (-NH-NH-) are bridges. For series A (directly linked) and series C (-N=N-), the chemical bonds in substitution groups become destroying bonds. Overall, it can indicate that the different bridges have a great influence on the determination of trigger bonds. Chung et al. [39] proposed that the BDE of a stable HEDM compound should be over the barrier 83.60 kJ/mol which means that almost all of the designed compounds possess suitable thermal stability (except for A3, A8, C3, C8, D2, D3, D4, D6).
Electrostatic potential
According to previous studies [23, 40, 41], there is a connection between the sensitivities of energetic compounds and the electrostatic potential (ESP) that is on the surface of the molecule. For ordinary organic molecules, the positive electrostatic potential will cover a larger area than the negative potential, but the latter tends to be a little stronger. However, in the case of the introduction of energetic groups (e.g., -C(NO2)3, -NO2) into the molecule, the positive potential will be strengthened and the negative potential will be weakened, so that the dominant position may become a positive potential.
Figure 4 shows the ESP and surface area of representative designed compounds (series D), where the ESP is calculated based on an electron density of 0.001 a.u. (election/Bohr3) and a lattice spacing of 0.25 Bohr. The local maximum and minimum values of the significant surface in the figure are shown in red and blue, respectively. It was observed that the red region (positive potential) was mainly concentrated on the parent, while the blue region (negative potential) was mainly distributed at the edges of the molecule, represented by the carbon atom on the parent oxadiazole and the oxygen atom on the nitro. The positive potential areas of the D1–D8 compounds were calculated as 149 Å2 (ratio 61%), 114 Å2 (ratio 54%), 145 Å2 (ratio 55%), 136 Å2 (ratio 58%), 153 Å2 (ratio 48%), 249 Å2 (ratio 68%), 117 Å2 (ratio 50%), and 165 Å2 (ratio 62%). Previous studies [42] reported that relatively sensitive molecules have regions of high electron indeed on covalent bonds throughout the molecular internal backbone, which are shown on the graph as a larger positive electrostatic potential on the surface area histogram. Easily, D1, D6, and D8 were the most sensitive. However, after comparing the electrostatic potential of the four series, it can be found when the substituent groups of the four series are the same, the positive potential areas of directly connected (A series) and the C and D series are not much different, while B series are slightly lower. And the comparison results also correspond to the results of the previous HOF and detonation performance. Detonation performances of the A, B, and C series are superior, the sensitivities are larger, and the situation is the opposite for the D series.


Electrostatic potential of representative designed molecules
Conclusions
In this work, we have designed 32 energetic materials based on 1,2,5-oxadiazole and 1,3,4-oxadiazole calculated their energetic properties and stability at the B3LYP/6-311 g (d,p) level, resulting in the following conclusions:
-
1.
Energetic groups play an important role in increasing the energy of HOMO and LUMO. When -NH2 and -NHNH2 are inserted, it contributes to the energy of HOMO, LUMO greatly.
-
2.
-N3 and -NH2, two nitrogen-containing groups, contribute to the HOF, especially the group of -N3, which contributes the most to the HOF. For the bridge, the different links do not have a significant impact on HOFs.
-
3.
When a single bond (directly linked) is bridged, the designed compound has a poor detonation performance.
-
4.
After analysis, most of the trigger bonds of the B series (-C=C-) are chemical bonds linked to the parent and substituent groups, the bonds of A series and C series are in the energetic groups.
Overall, -C(NO2)3 is the most effective strategy to improve the detonation performance of the design compounds (A6, D = 9.41 km s−1, P = 41.86 GPa, Q = 1572.25 cal g−1). Except for B4, B2, C2, D2, A2, and B1, the detonation velocities and detonation pressures of all the design compounds were higher than those of the conventional energetic material TNT.
In summary, the designed compounds are forecast to have outstanding detonation properties and affordable sensitivity, information that would probably prove valuable to researchers interested in these compounds. The findings of this research should contribute to the development of other new high explosives designs and discoveries.
References
Thottempudi V, Gao H, Shreeve JN (2011) Trinitromethyl-substituted 5-nitro-or 3-azo-1, 2, 4-triazoles: synthesis, characterization, and energetic properties. J Am Chem Soc 133(16):6464–6471. https://doi.org/10.1021/ja2013455
Wu Q, Zhu W, Xiao H (2014) A new design strategy for high-energy low-sensitivity explosives: combining oxygen balance equal to zero, a combination of nitro and amino groups, and N-oxide in one molecule of 1-amino-5-nitrotetrazole-3 N-oxide. J Mater Chem A 2(32):13006–13015. https://doi.org/10.1039/C4TA01879F
Sikder AK, Sikder N (2004) A review of advanced high performance, insensitive and thermally stable energetic materials emerging for military and space applications. J Hazard Mater 112(1–2):1–15. https://doi.org/10.1016/j.jhazmat.2004.04.003
Ma P, Wang J, Zhai D, Hao L, Ma C, Pan Y, Jiang J, Zhu S (2019) Structural transformation and absorption properties of 2, 4, 6-trinitro-2, 4, 6-triazacyclohexanone under high pressures. J Mol Struct 1196:691–698. https://doi.org/10.1016/j.molstruc.2019.07.020
Ma P, Pan Y, Jiang JC, Zhu SG (2018) Molecular dynamic simulation and density functional theory insight into the nitrogen rich explosive 1, 5-diaminotetrazole (DAT). Procedia Eng 211:546–554. https://doi.org/10.1016/j.proeng.2017.12.047
Sheremetev AB (1995) Chemistry of furazans fused to five-membered rings. J Heterocyclic Chem 32(2):371–385. https://doi.org/10.1002/jhet.5570320201
Pivina TS, Sukhachev DV, Evtushenko AV, Khmelnitskii LI (1995) Comparative characteristic of energy content calculating methods for the furazan series as an example of energetic materials. Propellants Explos Pyrotech 20(1):5–10. https://doi.org/10.1002/prep.19950200103
Sheremetev AB, Mantseva EV (1996) One-pot synthesis of 4, 4′-diamino-3, 3′-bifurazan. Mendeleev Commun 6(6):246–247. https://doi.org/10.1070/MC1996v006n06ABEH000745
Zhang C (2006) Computational investigation of the detonation properties of furazans and furoxans. THEOCHEM J Mol Struct 765(1–3):77–83. https://doi.org/10.1016/j.theochem.2006.03.007
Tang Y, He C, Mitchell LA, Parrish DA, Jean’Ne MS (2015) Energetic compounds consisting of 1, 2, 5-and 1, 3, 4-oxadiazole rings. J Mater Chem A 3.46 (2015): 23143–23148. https://doi.org/10.1039/C5TA06898C
Wang Q, Shao Y, Lu M (2019) Azo1,3,4-oxadiazole as a novel building block to design high-performance energetic materials. Cryst Growth Des 19(2):839–844. https://doi.org/10.1021/acs.cgd.8b01404
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X (2016). Gaussian:16
Fan XW, Ju XH (2008) Theoretical studies on four-membered ring compounds with NF2, ONO2, N3, and NO2 groups. J Comput Chem 29(4):505–513. https://doi.org/10.1002/jcc.20809
Wei T, Zhu W, Zhang X, Li YF, Xiao H (2009) Molecular design of 1, 2, 4, 5-tetrazine-based high-energy density materials. J Phys Chem A 113(33):9404–9412. https://doi.org/10.1021/jp902295v
Pan Y, Li J, Cheng B, Zhu W, Xiao H (2012) Computational studies on the heats of formation, energetic properties, and thermal stability of energetic nitrogen-rich furazano [3, 4-b] pyrazine-based derivatives. Comput Theor Chem 992:110–119. https://doi.org/10.1016/j.comptc.2012.05.013
O’Malley R (1983) Physical chemistry. J Chem Educ 60(2):A63. https://doi.org/10.1021/ed060pA63.2
Politzer P, Ma Y, Lane P, Concha MC (2005) Computational prediction of standard gas, liquid, and solid-phase heats of formation and heats of vaporization and sublimation. Int J Quantum Chem 105(4):341–347. https://doi.org/10.1002/qua.20709
Byrd EF, Rice BM (2006) Improved prediction of heats of formation of energetic materials using quantum mechanical calculations. J Phys Chem A 110(3):1005–1013. https://doi.org/10.1021/jp0536192
Lu T, Chen F (2012) Multiwfn: a multifunctional wave function analyzer. J Comput Chem 33(5):580–592. https://doi.org/10.1002/jcc.22885
Politzer P, Martinez J, Murray JS, Concha MC, Toro-Labbe A (2009) An electrostatic interaction correction for improved crystal density prediction. Mol Phys 107(19):2095–2101. https://doi.org/10.1080/00268970903156306
Kamlet MJ, Jacobs SJ (1968) Chemistry of detonations. I. A simple method for calculating detonation properties of C–H–N–O explosives. J Chem Phys 48(1):23–35. https://doi.org/10.1063/1.1667908
Lin H, Zhu Q, Huang C, Yang DD, Lou N, Zhu SG, Li HZ (2019) Dinitromethyl, fluorodinitromethyl derivatives of RDX and HMX as high energy density materials: a computational study. Struct Chem 30(6):2401–2408. https://doi.org/10.1007/s11224-019-01366-1
Stobiecka A, Sikora M, Bonikowski R, Kula J (2016) An exploratory study on the peroxyl-radical-scavenging activity of 2, 6-dimethyl-5-hepten-2-ol and its heterocyclic analogues. J Mol Struct 1107:82–90. https://doi.org/10.1016/j.molstruc.2015.11.043
Zhai D, Ma C, Ma P, Pan Y, Hao L, Liu X, Jiang J (2021) Theoretical insight into different energetic groups on the performance of energetic materials featuring RDX ring. Fuel 294:120497. https://doi.org/10.1016/j.fuel.2021.120497
Politzer P, Murray JS (2017) High performance, low sensitivity: the impossible (or possible) dream? Energetic materials. Springer, Cham, pp 1–22
Qu Y, Babailov SP (2018) Azo-linked high-nitrogen energetic materials. J Mater Chem A 6(5):1915–1940. https://doi.org/10.1039/C7TA09593G
Gutowski KE, Rogers RD, Dixon DA (2007) Accurate thermochemical properties for energetic materials applications. II. Heats of formation of imidazolium-, 1, 2, 4-triazolium-, and tetrazolium-based energetic salts from isodesmic and lattice energy calculations. J Phys Chem B 111(18):4788–4800. https://doi.org/10.1021/jp066420d
Miroshnichenko EA, Kon’kova TS, Matyushin YN (2003) Thermochemistry of primary nitramines. Dokl Phys Chem 392:253–255. https://doi.org/10.1023/A:1026182227885
Zhai D, Wang J, Hao L, Ma C, Ma P, Pan Y, Jiang J (2019) Molecular design and properties of bridged energetic pyridines derivatives. RSC Adv 9(65):37747–37758. https://doi.org/10.1039/C9RA07087G
Furka Á (2009) Relative energy of organic compounds II. Halides, nitrogen, and sulfur compounds. Struct Chem 20(4):605–616. https://doi.org/10.1007/s11224-009-9450-z
Mader CL (2007) Numerical modeling of explosives and propellants. CRC press
Wu Q, Pan Y, Xia X, Shao Y, Zhu W, Xiao H (2013) Theoretic design of 1, 2, 3, 4-tetrazine-1, 3-dioxide-based high-energy density compounds with oxygen balance close to zero. Struct Chem 24(5):1579–1590. https://doi.org/10.1007/s11224-012-0190-0
Yu Q, Wang Z, Wu B, Yang H, Ju X, Lu C, Cheng G (2015) A study of N-trinitroethyl-substituted aminofurazans: high detonation performance energetic compounds with good oxygen balance. J Mater Chem A 3(15):8156–8164. https://doi.org/10.1039/C4TA06974A
Hao L, Liu X, Zhai D, Qiu L, Ma C, Ma P, Jiang J (2020) Theoretical studies on the performance of HMX with different energetic groups. ACS Omega 5(46):29922–29934. https://doi.org/10.1021/acsomega.0c04237
Henry DJ, Parkinson CJ, Mayer PM, Radom L (2001) Radom, Bond dissociation energies and radical stabilization energies associated with substituted methyl radicals. J Phys Chem A 105.27: 6750–6756. https://doi.org/10.1021/jp010442c
Rice BM, Sahu S, Owens FJ (2002) Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules. THEOCHEM J Mol Struct 583(1–3):69–72. https://doi.org/10.1016/S0166-1280(01)00782-5
Harris NJ, Lammertsma K (1997) Ab initio density functional computations of conformations and bond dissociation energies for hexahydro-1, 3, 5-trinitro-1, 3, 5-triazine. J Am Chem Soc 119(28):6583–6589. https://doi.org/10.1021/ja970392i
Lu T, Chen F (2013) Bond order analysis based on the Laplacian of electron density in fuzzy overlap space. J Phys Chem A 117(14):3100–3108. https://doi.org/10.1021/jp4010345
Chung G, Schmidt MW, Gordon MS (2000) An ab initio study of potential energy surfaces for N8 isomers. J Phys Chem A 104(23):5647–5650. https://doi.org/10.1021/jp0004361
Murray JS, Concha MC, Politzer P Links between surface electrostatic potentials of energetic molecules, impact sensitivities and C–NO2/N–NO2 bond dissociation energies. Mol Phys 107.1: 89–97. https://doi.org/10.1080/00268970902744375
Peter JS (1998) Effects of strongly electron-attracting components on molecular surface electrostatic potentials: application to predicting impact sensitivities of energetic molecules. Mol Phys 93(2):187–194. https://doi.org/10.1080/002689798169203
Rice BM, Hare JJ (2002) A quantum mechanical investigation of the relation between impact sensitivity and the charge distribution in energetic molecules. J Phys Chem A 106(9):1770–1783. https://doi.org/10.1021/jp012602q
Availability of data and material
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
Funding
This study was supported by the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 20KJB620001) and the National Natural Science Foundation of China (Grant No. 11702129).
Author information
Authors and Affiliations
Contributions
Wenxin Xia: software, data curation, writing—original draft preparation, formal analysis; Congming Ma: conceptualization, methodology, Peng Ma: resources, project administration; Renfa Zhang: investigation, formal analysis; Xiaosong Xu: investigation, validation; Yong Pan: resources; Juncheng Jiang: resources, supervision
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 1742 kb)
Rights and permissions
About this article

Cite this article
Xia, W., Zhang, R., Xu, X. et al. Theoretical studies of novel high energy density materials based on oxadiazoles. J Mol Model 27, 204 (2021). https://doi.org/10.1007/s00894-021-04805-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-021-04805-1