
Abstract
The quantification of bond strengths is a useful and general concept in chemistry. In this work, a Coulombic force model based on atomic electric charges computed using the accurate distributed multipole analysis (DMA) partition of the molecular charge density was employed to quantify the weakest N–NO2 and C–NO2 bond strengths of 19 nitrobenzene, 11 nitroazole, and 10 nitramine molecules. These bonds are known as trigger linkages because they are usually related to the initiation of an explosive. The three families of explosives combine different types of molecular properties and structures ranging from essentially aromatic molecules (nitrobenzenes) to others with moderate aromaticity (nitroazoles) and non-aromatic molecules with cyclic and acyclic skeletons (nitramines). We used the results to investigate the impact sensitivity of the corresponding explosives employing the trigger linkage concept. For this purpose, the computed Coulombic bond strength of the trigger linkages was used to build four sensitivity models that lead to an overall good agreement between the predicted values and available experimental sensitivity values even when the model included the three chemical families simultaneously. We discussed the role of the trigger linkages for determining the sensitivity of the explosives and rationalized eventual discrepancies in the models by examining alternative decomposition mechanisms and features of the molecular structures.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The concept of chemical bond is central to chemistry. Therefore, different theoretical models, especially the molecular orbital (MO) and valence bond (VB) approaches, have been employed successfully to predict structure, stability, and chemical reactivity of molecules, difficult properties to investigate experimentally [1, 2]. Other successful bonding models are based on the Ruedenberg idea that covalent bonds are due to quantum interference between one-electron states [3, 4] or on an electron density model such as Bader’s Atoms in Molecules (AIM) [5].
Considering that the chemical bond is only a theoretical construct [6], all bond properties (e.g., bond strengths) “can only be defined within models and thus are not measurable” [2]. For this reason, accurate theoretical calculations based on sound bonding models can provide insights into the nature and properties of different types of interactions [7], hence on relevant chemical properties. For instance, several groups employ the bond dissociation energy (BDE) to measure bond strength for investigating the sensitivities of energetic materials and other properties [8,9,10,11,12,13].
Therefore, the possibility of quantifying bond strengths employing different approaches can enlighten a variety of chemical phenomena. In this work, we employed a Coulombic bonding model [14, 15] to quantify specific bond strengths to study the impact sensitivity of nitrobenzene, nitroazole, and nitramine explosives.
Explosives, propellants, and pyrotechnics are controllable storage systems of chemical energy having important technology roles [16]. These energetic materials (EMs) are primarily molecular solids composed of polyatomic organic molecules bearing complicated crystal structures [17,18,19]. Nitramine, nitrobenzene, and nitroazole explosives are characterized by N–NO2 and C–NO2 bonds, combining relative insensitivity to initiation and great energetic density [20,21,22].
Some authors have been concerned with determining molecules’ tendency to break and form bonds from electronic and topological properties computed throughout a reaction process [23]. However, our focus is in the initiation process of the decomposition of energetic materials, particularly on the specific bonds that when broken, initiate the entire process of decomposition of the explosive. Once determined how easy it is to cleave this bond, the tendency to initiate the explosive process can be evaluated.
The most useful EMs should have low sensitivity to prevent an accidental explosion by shock, impact, friction, heat, or electric sparks during transportation or storage [24,25,26]. On the other hand, a high level of performance of an EM usually is on equal footing with an enhanced sensitivity [27, 28]. In other words, an insensitive explosive, in general, does not exhibit good performance. These apparently incompatible requirements also imply the necessity of comprehensive knowledge of the sensitivity of EMs.
We concentrate here on impact as a stimulus because this property has some correlation within families of EMs [28]. Impact sensitivity can be quantified by measuring h50, which is the height from which a given standard mass dropped upon a sample of the compound will initiate the reaction 50% times; naturally, h50 depends also on the dropping mass [29,30,31,32,33]. An in-depth discussion on the drop-weight impact sensitivity test as a tool for measuring the impact sensitivity of explosives has just been published [34].
Although the measurement of sensitivity by impact tests has different sources of uncertainty, this property correlates with chemical structure [35,36,37] because the sensitivity of EMs is basically determined by the chemical character of materials [27]. For this reason, computing molecular properties via accurate quantum chemical methods to correlate them with sensitivity values often has been successful [38,39,40,41].
It is believed that the bond strength of the weakest bond in the molecule of an EM, the trigger (bond) linkage, is crucial for initiation events [12]. In nitro-containing explosives, C–NO2, N–NO2, and O–NO2 bonds would be the trigger linkages [11].
The idea of relating the positive electrostatic potential of trigger linkages to BDEs (hence to bond strengths) and sensitivity for several high (secondary) explosives bearing C–NO2 and N–NO2 bonds has been very fruitful [29, 42,43,44,45]. It was found overall that correlation between bond strengths and impact sensitivity is not limited to certain classes of explosives [16]; the correlation with the reciprocals of bond lengths (another measure of bond strength) was also investigated [46].
From the computed C–NO2 and N–NO2 BDEs, a correlation was found between h50 and the BDE of the weakest bond [47,48,49]. Fried et al. introduced the BDE/Ed ratio, where Ed is the energy content, to derive relationships between this ratio and impact sensitivity [16], later modified by using instead the total energy of the molecule (Etotal) computed from density functional theory (DFT) methods [11, 50]. A relationship between nitro compounds and nitramines’ sensitivity with energy of dissociation and energy content/molecular energy per atom was found [51, 52]. Keshavarz and coauthors explored the correlation of molecular structural parameters of different families of EMs with impact sensitivity [53,54,55,56,57].
Another approach evaluated the trigger bond strengths of various explosives, especially nitroaromatics, using a relative Wiberg bond index (WBI) [58]. It was found that WBIs would be a better predictor of trigger bond strength compared to BDEs.
Inspired in those works, we have been employing the accurate distributed multipole analysis (DMA) partition scheme of the molecular charge densities developed by Stone [59,60,61,62] to investigate impact sensitivity and decomposition of diazocyclopropanes [63], nitraromatics [64, 65], RDX (hexahydro-1,3,5-trinitro-1,3,5-triazine) [66], and FOX-7 (1,1-diamino-2,2-dinitroethylene) molecules [67]. We recently proposed DMA mathematical models of sensitivity for 14 cyclic nitramine [68] and 33 nitroazole [69] molecules. This partition method was employed before to rationalize hydrodesulfurization (HDS) catalytic processes [70,71,72] and optimum cluster size for adsorption investigations [73].
Those efforts attempted to correlate the bulk property impact sensitivity with the molecular properties of EMs. However, there have been criticisms [74] claiming that within this approach dominating chemical mechanisms of initiation reactions can be obscured [75]. Therefore, it should not be used for interpretation, but instead for identifying molecular properties related to impact sensitivity [29]. This is our purpose here.
In this work, we developed sensitivity models based on a Coulombic bonding model employing DMA charges to compute bond strengths of the N–NO2 and C–NO2 trigger linkages of 10 nitramines, 11 nitroazoles, and 19 nitrobenzene molecules. The three families of explosives combine different types of molecular properties and structures.
Computational methods and theoretical background
We investigated the 10 nitramine, 11 nitroazoles, and 19 nitrobenzenic molecules depicted in Scheme 1. Table 1S of the Supplementary Material identifies the acronyms.



Structural formulas of the investigated molecules
To determine each molecule’s lowest energy geometry, a conformational search was initially carried out employing a Monte Carlo method combined with simulated annealing and DFT//SVWN/DN*. Different conformers were generated by randomly rotating bonds and bending rings to produce diverse geometries. The Spartan Pro software [76] was used for this task. The resulting structures were the starting point for determining the lowest energy conformer in each case.
The geometry of all molecules found in that way and the corresponding molecular charge densities were further optimized with the DFT/B3LYP//6–311 + G(d) method [77,78,79] employing the Gaussian 03 package [80]. Vibrational frequency calculations confirmed that each converged geometry corresponds to a minimum on the potential energy surface. The lowest energy conformer was used to compute the DMA. For the nitrobenzene molecules, single-point DFT/B3LYP//6–311 + G(d) calculations were performed on published (M06-2X)//TZVP geometries [58].
The charge density of each molecule’s lowest energy geometry was partitioned employing the DMA implemented in the GDMA2 program [62]. The DMA expansion furnishes a very accurate description of the molecular charge density. It is a quantitative population analysis with a sound physical-chemical foundation providing straight chemical interpretability [59].
The DMA method divides the charge density into a sum of a product of atom-centered Gaussian basis functions; the coefficient of each term is determined from the one-electron density matrix [59,60,61]. A basis function product is then written as a sum of electric multipole moments of ranks up to the degree of its polynomial. In this way, the overlap of two s functions represents a pure monopole (charge), the product of s with a p corresponds to a monopole plus a dipole, two p functions when overlapping produces charge, dipole and quadrupole moments; higher electric multipole terms can be generated so the process can go on.
When the orbitals are localized on different atoms, a given pair of Gaussian functions generates a finite multipole series at a point between the two atoms: the exponents of the Gaussian orbitals determine the exponent of this point. Thus, the multipoles are written as a series localized on the nearest atom. The DMA approach computes these exact representations approximating each one by a multipole expansion, usually centered on the atomic nuclei.
The series produced by the DMA rapidly converges because of the expansion on different points of the electronic charge distribution. Upon combining the decomposed charge distribution with the nuclei’s positive charges, an accurate representation of the molecular charge density is obtained.
The DMA expansion terms have a direct chemical interpretation. The monopole (charge) term represents charges on atomic sites, with bonds between adjacent atoms usually having some degree of charge separation. Dipole and quadrupole electric moments also have a direct interpretation [81]. We used here only the DMA charge values.
To compute the N–NO2 and C–NO2 bond strengths, a Coulombic bonding model previously used [68, 71] was employed. This model considers that bond strength values computed by the atom-centered DMA expansion up to the quadrupole term are dominated by the Coulombic interactions between DMA charges in each atom of the bonding pair.
The molecular charge density is a property that includes information on the molecular bonds. Given the DMA partition of the charge density, bonding properties are built-in in the atom-centered electric multipole moments. Therefore, the computed bond strengths provided by the Coulombic force values computed from the (DMA charge)−(DMA charge) products include, albeit indirectly, the covalent contribution to the bond strength.
Furthermore, those values of bond strengths are accurate because the DMA charge−DMA charge contribution largely dominates the interaction between atom-centered electric multipoles involved in a chemical bond: the ion-dipole, dipole-dipole, and higher order interaction energies are much smaller as compared with the ion–ion term. Numerically, ion-ion interaction energies are ~17 greater than ion-dipole interaction energies, ~125 greater than dipole-dipole interaction energies, and smaller for higher-order terms—see Table 18.3 in Ref. [82]. In other words, for the DMA partition of the molecular charge density, the largest contribution to the computed Coulombic bond strength values is by far due to the charge-charge Coulombic interaction. This fact does not mean that the corresponding bonding is ionic, but just that the magnitudes of the bond strengths computed in this way are dominated by the (DMA charge)−(DMA-charge) values.
Therefore, using a Coulombic model to compute bond strengths combines (a) reliable quantitative and semi-quantitative information on bond strengths, as pointed out before [14, 15]; (b) the possibility of rationalizing other chemical phenomena as we have done [71,72,73]; and (c) the use of the accurately computed DMA charges as input.
From the well-known expression of the Coulomb’s Law, the magnitude of the Coulombic force F of the N–NO2 and C–NO2 bonds is given by
where F is the magnitude of the Coulombic force between the N–N and C–N atoms of the trigger bonds, q1 and q2 are the corresponding DMA monopole (charge) values in units of elementary charge e (1.602 × 10−19 C), and R is the N-NO2 or C–NO2 bond lengths in Å. We took the SI constant 1/4πε0 of the usual form of the Coulomb’s Law as equal to one.
The computed F values were used to quantify the magnitude of the N–NO2 and C–NO2 bond strengths. For this purpose, we define the normalized weakest N–NO2 and C–NO2 Coulombic force Fnw, which is the weakest force in a molecule thus is the trigger linkage, according to the following expressions:
where [#NNO2]2 and [#CNO2]2 are respectively the square of the sum of the number of the N–NO2 and C–NO2 bonds. These sums quantify the number of moieties that determine the explosive behavior of the nitramines, nitroazoles, and nitrobenzenes, and in a certain sense, indicate the molecular size. We found here that squaring this quantity, instead of just using the bare number, gives much better results: this is probably due to a non-linear prominent explosophore roles of the N–NO2 and C–NO2 bonds, and their increased importance in this regard the larger their number. Politzer, Murray, and coworkers 30 years ago, in a paper investigating the sensitivity of nitroamines and nitroaliphatics, noticed that a good correlation could be obtained by including the number of N–NO2 linkages given that “their effectiveness in triggering decompositions is “diluted” as the molecule becomes larger” [83]. In our case (Eqs. 2a and 2b), this “dilution effect” appears as the square of the sum of the number of linkages.
The Coulombic force equals minus the gradient of the electrostatic potential: F = − ∇ V(r). Hence, the former captures variations of V(r), which is the integrated charge density [84]. V(r) has been used to investigate impact sensitivity (e.g.; Ref. [44]) and noncovalent interactions of halogen complexes [85, 86], among a variety of other chemical properties.
In a nice and comprehensive work on the Hellmann-Feynman theorem, Politzer and Murray elucidated the fact that “in physical reality the forces felt by the nuclei and molecules and other systems are entirely Coulombic” and this physical fact does not depend on the approximate nature of a quantum chemical wave function or electronic density [87]. This important and not well-known fact motivates the present effort.
Considering a given computed molecular charge distribution and its realistic atom-centered DMA representation, the use of DMA charges could lead to accurately quantitative Coulombic force values as a measure of bond strengths. The results discussed below show that.
Results and discussion
Initially, to design a general model for determining h50, all 40 molecules were considered, regardless of the families to which they belong. Therefore, the complete set of molecules was divided into two groups. The training group included 19 molecules chosen according to a diversity of structures, group of atoms and type of bonds. The remaining 21 molecules comprised the test group used to verify the prediction of the model.
Fig. 1S of the Supplementary Material collects the computed bond distances and DMA charge values, employed to determine the magnitude of the weakest normalized Coulombic Force (Fnw) of all 40 molecules, as defined by Eqs. (2). The computed Fnw values are shown in Table 2S.
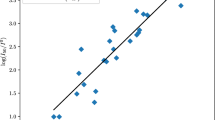
The plot of Fnw versus the experimental h50 values for the 19 molecules of the training group is shown in Fig. 1.

Correlation between the experimental h50 values and the normalized weak force Fnw for the 19 molecules of the training group
A linear model for the sensitivity was then derived from Fig. 1:
where the units of the fitted constants are respectively cm.Å2e−2 and cm. The results computed from Eq. 3 are shown in Table 1.
Overall, for the three families of explosives, there is a good correlation between the weak normalized force Fnw and the experimental h50 values (R2=0.9545), hence between the trigger linkage strength computed from DMA charges and the measured sensitivity values. This model is quite general in the sense that predicts the sensitivity of explosives considering that the three families of explosives bear different chemical properties and two types of trigger bonds.
From Table 1, it can be seen that 15 molecules have deviations below 20%. This is a quite good predictability considering such a heterogeneous group of molecules.
Four molecules display large deviations of h50 from the predicted values: CL20 (231%), TNCB (369%), MEDINA (788%), TNTRICB (184%). These molecules are nitramines, in contrast with the aromatic character of most of the others, which might explain the large discrepancy.
Would these already good results improve if the three families of explosives were treated separately, especially for the most discrepant results? We answer this question in the following three subsections, selecting for each family as the training group the same molecules of each family employed in the training group of the general model given by Eq. 3.
Nitramines
Figure 2 plots Fnw vs. h50 of the four nitramine molecules of the training group.

Correlation between the impact sensitivity (h50) and the weakest normalized N-NO2 bond strength Fnw of the nitramine molecules of the training group
Even considering the small number of molecules in the training group, there is a good correlation between the strength of the weakest normalized N–NO2 bond and the impact sensitivity of the four analyzed nitramines (R2 = 0.9432). From the linear fitting in Fig. 2, the following linear correlation between Fnw and h50 was found:
where the fitted constants have the same units of Eq. 3.
Equation 4 was employed to predict the h50 values of the remaining 10 nitramine molecules. A comparison with available experimental data is shown at Table 2.
Considering the group of nitramines separately, out of the 10 molecules, 3 have deviations below 20% (CL20, DINGU, and OCPX). On the other hand, MEDINA (710%), NQ (112%), and TNCB (118%) displayed the largest deviations. The deviations of CL20 and TNTRICB greatly decreased as compared with the general model, given by Eq. 3, by respectively from 231→0% and 184→43%. In the following, we discuss the possible reasons for the large deviation of the results for the MEDINA, NQ, and TNCB molecules.
MEDINA is one of the three acyclic molecules in the set of nitramines and differs from OCPX only for the lack of methyl groups attached to the amine nitrogen atom—see Scheme 1. However, this structural difference is sufficient for MEDINA to have a much larger deviation and sensitivity (710% and h50 = 114 cm) compared with OCPX (10% and h50 = 13 cm) according to the model described by Eq. 4.
MEDINA is a primary nitramine, in contrast with OCPX that is a secondary nitramine; therefore, it decomposes differently. The products of both thermal and in aqueous solution decomposition of MEDINA consist mainly of nitrous oxide and formaldehyde, as is depicted in the reaction below [92]:
Therefore, the decomposition of MEDINA molecules does not start with the cleavage of the N–NO2 bond, as assumed by our model. In fact, the mechanism proposed a few decades ago indicates the initial cleavage of the C–N bond after an electronic delocalization process [92]:

Hence, the trigger linkage in MEDINA is other than the one assumed here. Additionally, considering that the rupture of that trigger linkage is driven by an electronic rearrangement in the molecule, and not because the N–NO2 bond is the weakest, this behavior might explain the large deviation in our model. This mechanism is also confirmed by the fact that the normalized Coulombic force Fnw of the C–N bond (0.148 e2 Å−2) is more than four times weaker as compared with the Fnw value of the N–N bond (0.605 e2 Å−2) used as input in our models.
The non-cyclic nitroguanidine (NQ) explosive has the highest experimental h50 value of the set (>320 cm). This fact was reproduced by Eqs. 3 and 4, despite the considerable percentual errors in both cases, respectively 76.8% and 112%. In both models, NQ is highly insensitive because of the substantial value of the normalized Coulombic force Fnw (3.93 e2 Å−2) of the N–NO2 bond compared with the forces of the other bonds—this is the largest computed Fnw force value in the whole set of 40 molecules (see Table 2S of the Supplementary Material).
For the NQ molecule, both intramolecular and intermolecular hydrogen bonds can be formed. The existence of hydrogen bonding in NQ also leads to the formation of a stable six-membered cyclic arrangement with an extensive electron-delocalized structure (Fig. 3), which contributes to stabilize the molecule [93] thereby decreasing its sensitivity.

Intramolecular hydrogen interaction in NQ and its extensive electron-delocalized structure
Still considering the NQ molecule, it is possible to identify two possible structures (Fig. 4).

Nitramine (a) and nitrimine (b) structures for NQ
NQ has been studied for decades in order to determine precisely its structure [94]. It is now accepted that NQ, both crystalline and in solution, is in the form of nitrimine (Fig. 4b). Therefore, in gas-phase, it is expected to have the same conformation. However, although there is an agreement concerning its structure, it is oddly observed that the length of the iminic C–N bond is larger as compared with the C-NH2 separation; thus, the former is a weaker bond. Efforts have been made to refine these bond lengths and to resolve this apparent contradiction [94].
The converged geometry (Fig. 5) shows what we should really expect from the bond length of a nitrimic structure in NQ, that is, a shortened C=NNO2 bond in comparison with the other aminic C-NH2 bonds. Considering that DMA monopole values depend on the molecular geometry, and given that our optimized geometry differs from the actual one, the predicted sensitivity values based on the DMA charges are different from the experimental values. This is a possible reason for the large deviation found for this molecule.

Optimized geometry of NQ showing some bond lengths
Due to regions of planarity in the TCNB molecule (Fig. 6), there is a repulsion between the non-bonding electron pairs of the oxygen atoms of the nitro groups and the π bond of the carbonyl group. Shoaf and Bayse concluded that this repulsive interaction between the clouds of isolated electron pairs in very electronegative atoms is responsible for a remarkable instability of this explosive displaying this spatial arrangement [58]. This is probably a strong reason for the large deviation.

Molecular structures of TNTriCB (a) and TNCB (b) evidencing different arrangements of the nitro groups
Furthermore, the resonance structures observed for the carbonyl-nitramine system in TNCB proposed in Fig. 7 suggest an additional possibility. This electronic delocalization favors the disruption of the OC–NNO2 bond over the more common N–NO2 bond rupture mechanism of our models, a behavior that could increase the degree of error in the predicted h50 value.

Proposed resonance structures for a region of the TNCB molecule: a the amine nitrogen electron pair; b and c: the nitro and the carbonyl groups and the electron pair of the amine nitrogen
In particular, the resonance structures in Fig. 7(b) and (c) are responsible for disrupting the C–N bond instead of the N–N bond in TNCB. The normalized Coulombic force of the C–N bond is 0.136 e2Å−2 in contrast with 0.133 e2Å−2 of the N–N bond. Therefore, since the strength values of the two bonds are similar, the aforementioned electronic delocalization really allows the C–N bond to function as the trigger linkage of the molecule. Moreover, for TNTriCB, which does not have this resonance structure in spite of having a similar backbone geometry, a large deviation was not found.
Comparing the present results with our previous work that developed impact sensitivity models employing other DMA multipole values [68], there is here an overall improvement. Considering a set of 10 molecules, 7 showed results closer to experiment as compared with the previous work. Therefore, the Coulombic force model using DMA charges to compute bond strengths of trigger linkages provides an improved description of the impact sensitivity of nitramine explosives.
Nitroazoles
Figure 8 plots Fnw vs. h50 for the 5 nitroazole molecules of the training group having experimental h50 values.

Correlation between the impact sensitivity (h50) and the weakest normalized C-NO2 bond strength Fnw of the nitroazole molecules of the training group
A good correlation was found (R2 = 0.9728). The mathematical model for sensitivity derived from the curve in Fig. 8 is
where the units for the fitted constants are respectively cm.Å2e−2 and cm.
The results computed from Eq. 5 are collected on Table 3.
For nitroazoles, the fraction of molecules with very satisfactory deviations is larger compared with the nitramines. Here, six molecules (ANTA, 24DNI, 3N124TRIAZ, NTO, 34DNPY, and 1MET35DNI124TRIAZ) have deviations below 20%.
The largest, though not very large, deviations were found for the LLM116 (44%) and 35DNPY (40%) molecules. In LLM116, two nitro groups are intercalated by an amino group (NH2). The presence of this moiety between the nitro groups allows the formation of an intramolecular hydrogen bond. In a similar way, in 35DNPY, the hydrogen bond is between the oxygen of the nitro group and the hydrogen in position 1, as shown in Fig. 9.

Intramolecular hydrogen bonds of the LLM-116 and 35DNPY molecules
Therefore, the formation of two six-membered rings in LLM-116 and of a five-membered ring in 35DNPY is possible (Fig. 9). Since these rings have good stability, the rupture of the C–NO2 trigger linkage is more hindered, leading to a large h50 value (167.50 cm and 168 cm, respectively). In spite of this, a reasonable deviation between the predicted and the experimental values of h50 was found (44% for LLM-116 and 40% for 35DNPY). The influence of hydrogen bonds on the bond strengths of LLM-116 is also confirmed by other theoretical studies [101,102,103].
Nitrobenzenes
For the nitrobenzene family, a plot of the measured values of Fnw vs. h50 for the training group is depicted in Fig. 10.

Correlation between the weakest normalized bond force Fnw of the molecules of the training group and the impact sensitivity (h50) of the nitrobenzene molecules
From the training group with 10 molecules, the linear fitting results in:
where the units of the fitted constants are respectively in cmÅ2e−2 and cm units.
The results obtained for the 19 nitrobenzene molecules employing Eq. 6 are shown in Table 4. The correlation in this case (R2 = 0.8049) is not as good as for the two previous families.
For the nitrobenzenes, the largest group, nine molecules (24DNP, HNBP, HNDPA, HNS, PA, DClTNAN, TMTNB, DMTNB, and TNAP) have deviations below 20%. Once again, the good result already observed for the other aromatic family (nitroazoles) and for the general model is reproduced here.
Overall, the deviations are not large. The worst results are for EOTNB (57%), HNDPM (58%), MATENT (81%), and TETNA (59%) (see Fig. 11 and Table 4).

Nitrobenzene molecules with the larges deviations resulting from the sensitivity prediction model of Eq. 6
Those deviations are probably related to the intramolecular transfer of a hydrogen atom, as showed before [58, 104,105,106]. When this transfer occurs, there are two different reaction mechanisms illustrated in Fig. 12 for the MATENT and TETNA molecules. The mechanism is the probable cyclization occurring between the nitro and the amino groups. When cyclization occurs, the bond between the carbon atom and the nitrogen atom of the nitro group becomes stronger hence is more difficult to break. Therefore, when this cycle is formed, the original trigger bond cannot be involved in the rupture process.

Possible cyclization mechanisms for the a TETNA and b MATETNT molecules
For MATETNT, according to the computed force values, the weakest bond is at position 2 and the strongest at position 5 (Figs. 11 and 12b). This result is inconsistent with the proposed mechanism in the literature since, when cyclization occurs, the bond at position 2 becomes stronger, and this can only occurs when the nitro group is at position 2 or 4. Therefore, it is plausible to imagine that the weaker bonds would be at positions 5 and 6.
Given the very similar structures, the analysis for the TETNA molecule is identical. The bonds involving carbons 2 and 6 should be the strongest, since it is possible to occur cyclization between the amino group located on carbon 1 and the nitro groups at positions 2 and 6 (Figs. 11 and 12a). The trigger linkages, therefore, could be at positions 3 and 4, since these should be the weakest in the structure. However, when calculating the corresponding forces, the weakest bond is actually at carbon 6, which is inconsistent with our force model, as it should be the strongest bond and not the weakest one.
Regarding biphenyl molecules, they are sensitive compounds due to steric effects and repulsion between nitro groups [58]. If we consider the HNDPM, HNS, and HNBP molecules, our model predicted decreasing values of sensitivity, which is consistent with experimental values (Table 4). Figure 13 shows the optimized structures of the three biphenyl molecules.

Optimized structures of the HNDPM, HNBP, and HNS molecules
According to Fig. 13, the aromatic rings of the HNDPM are close to each other, thereby increasing the repulsion between their nitro groups more than in the other two molecules (HNBP and HNS).
Considering EOTNB, a repulsive electronic effect between the oxygen atoms in OEt and the nitro group seems to play a determining role in the relatively large predicted deviation (57%). As pointed out before [58], these groups bearing non-bonded electron pairs localized in highly electronegative atoms interact repulsively with the electronegative nitro group. This effect could be the reason for the observed deviation.
Conclusions
We presented a concept of a Coulombic force of a bond, based on the DMA monopole (charge) values, as a measure of bond strength. The forces computed in this way were used to develop correlation models between impact sensitivity (given by h50 values) and force values. For this purpose, the trigger linkage concept, related to the decomposition initiation of several explosives, was used.
Molecules from three distinct chemical families of explosives were investigated. They were 11 non-aromatic nitramines, 10 nitroazoles, and 19 nitrobenzenes bearing different molecular structures and chemical properties.
We found a very good correlation between the weakest normalized force (Fnw), defined as the Coulombic force divided by the squared number of N–NO2 or C–NO2 bonds of each molecule, and the corresponding h50 value. The proposed mathematical model for describing this correlation proved to be sufficiently broad to predict the sensitivity values of the three explosive families considered simultaneously. Its application to the total set of 40 molecules led to predicted h50 values mostly having small relative deviations. The reasons of the large deviations found for the CL20, TNCB, TNTRICB, and MEDINA molecules were thoroughly discussed by examining different decomposition mechanisms and features of the molecular structures. The same force model was applied to each family of explosives separately to rationalize these differences.
Some large deviations in the three additional models, which considered the weakest normalized forces for each family separately, still persisted. However, for the following molecules, they were not as large as before: NCB, MEDINA, NQ, MATETNT, TETNA, HNDPM, and EOTNB. These deviations could be mostly associated mainly with specific decomposition mechanisms not involving a trigger bond (e.g.; MEDINA, TNCB, MATETNT and TETNA). In other cases, (HNDPM and EOTNB), they were due to the electronic repulsion and conformational particularities of their molecular structures.
In this work, we proposed a general model to compute bond strengths from a Coulombic model having as input the DMA charge (monopole) values. This model was employed successfully to describe the impact sensitivity of explosives composed of molecules from three different chemical families, either considered altogether or separately, which shows its generality. Other applications of this force model to different chemical phenomena are under way.
Data availability
All the data can be found in the Supporting Information.
References
McWeeny R, Coulson CA (1980) Coulson's Valence. Oxford University Press, Oxford
Cremer D, Wu AN, Larsson A, Kraka E (2000) Some thoughts about bond energies, bond lengths, and force constants. J Mol Model 6(4):396–412
de Sousa DWO, Nascimento MAC (2017) Are one-electron bonds any different from standard two-electron covalent bonds? Acc Chem Res 50(9):2264–2272
Fantuzzi F, Cardozo TM, Nascimento MAC (2017) On the metastability of doubly charged homonuclear diatomics. Phys Chem Chem Phys 19(29):19352–19359
Bader RFW (1994) Atoms in molecules: a quantum theory. International series of monographs on chemistry, vol 22. Clarendon Press, Oxford
Politzer P, Murray JS (2019) A look at bonds and bonding. Struct Chem 30(4):1153–1157
Romanova A, Lyssenko K, Ananyev I (2018) Estimations of energy of noncovalent bonding from integrals over interatomic zero-flux surfaces: correlation trends and beyond. J Comput Chem 39(21):1607–1616
Su XF, Cheng XL, Meng CM, Yuan XL (2009) Quantum chemical study on nitroimidazole, polynitroimidazole and their methyl derivatives. J Hazard Mater 161(1):551–558
Turker L, Atalar T (2006) Quantum chemical study on 5-nitro-2,4-dihydro-3H-1,2,4-triazol-3-one (NTO) and some of its constitutional isomers. J Hazard Mater 137(3):1333–1344
Jun Z, Xin-lu C, Bi H, Xiang-dong Y (2006) Neural networks study on the correlation between impact sensitivity and molecular structures for nitramine explosives. Struct Chem 17(5):501–507
Song XS, Cheng XL, Yang XD, Li DH, Linghu RF (2008) Correlation between the bond dissociation energies and impact sensitivities in nitramine and polynitro benzoate molecules with polynitro alkyl groupings. J Hazard Mater 150(2):317–321
Atalar T, Jungova M, Zeman S (2009) A new view of relationships of the N-N bond dissociation energies of cyclic nitramines. Part II. Relationships with Impact Sensitivity. J Energ Mater 27(3):200–216
Kraka E, Cremer D (2009) Characterization of CF bonds with multiple-bond character: bond lengths, stretching force constants, and bond dissociation energies. ChemPhysChem 10(4):686–698
Sacks LJ (1986) Coulombic models in chemical bonding: 1. Description and some applications of a Coulombic model. J Chem Educ 63(4):288–296
Sacks LJ (1986) Coulombic models in chemical bonding: 2. Dipole-moments of binay hydrides. J Chem Educ 63(5):373–376
Fried LE, Manaa MR, Pagoria PF, Simpson RL (2001) Design and synthesis of energetic materials. Annu Rev Mater Res 31:291–321
Kuklja MM, Stefanovich EV, Kunz AB (2000) An excitonic mechanism of detonation initiation in explosives. J Chem Phys 112(7):3417–3423
Dippold AA, Klapotke TM (2013) A study of Dinitro-bis-1,2,4-triazole-1,1 '-diol and derivatives: design of high-performance insensitive energetic materials by the introduction of N-oxides. J Am Chem Soc 135(26):9931–9938
Klapotke TM, Krumm B, Widera A (2018) Synthesis and properties of Tetranitro-substituted adamantane derivatives. ChemPlusChem 83(1):61–69
Badgujar DM, Talawar MB, Asthana SN, Mahulikar PP (2008) Advances in science and technology of modern energetic materials: an overview. J Hazard Mater 151(2–3):289–305
Trache D, Tarchoun AF (2018) Stabilizers for nitrate ester-based energetic materials and their mechanism of action: a state-of-the-art review. J Mater Sci 53(1):100–123
Sivabalan R, Anniyappan M, Pawar SJ, Talawar MB, Gore GM, Venugopalan S, Gandhe BR (2006) Synthesis, characterization and thermolysis studies on triazole and tetrazole based high nitrogen content high energy materials. J Hazard Mater 137(2):672–680
Nanayakkara S, Kraka E (2019) A new way of studying chemical reactions: a hand-in-hand URVA and QTAIM approach. Phys Chem Chem Phys 21(27):15007–15018
Politzer PM, J. S. (2003) Energetic materials. Part 1. Decomposition, crystal and molecular properties, vol 12. Theoretical and Computational Chemistry. Elsevier, Amsterdan
Politzer PM, J. S. (2003) Energetic materials. Part 2. Detonation, Combustion., vol 13. Theoretical and Computational Chemistry. Elsevier, Amsterdan
Shaw RWB, T. B., Thompson DL (2005) Overviews of recent research on energetic materials, vol 16. Advanced Series in Physical Chemistry, World Scientific, New Jersey
Zeman S, Jungova M (2016) Sensitivity and performance of energetic materials. Propellants Explos Pyrotech 41(3):426–451
Politzer P, Murray JS (2016) High performance, low sensitivity: conflicting or compatible? Propellants Explos Pyrotech 41(3):414–425
Rice BM, Hare JJ (2002) A quantum mechanical investigation of the relation between impact sensitivity and the charge distribution in energetic molecules. J Phys Chem A 106(9):1770–1783
Zeman S (2007) Sensitivities of high energy compounds. High energy density materials, vol 125. Structure and Bonding. Springer-Verlag, Berlin, Berlin, pp 195–271
Mathieu D (2013) Toward a physically based quantitative modeling of impact sensitivities. J Phys Chem A 117(10):2253–2259
Mathieu D, Alaime T (2014) Predicting impact sensitivities of nitro compounds on the basis of a semi-empirical rate constant. J Phys Chem A 118(41):9720–9726
Mathieu D (2015) Prediction of gurney parameters based on an analytic description of the expanding products. J Energ Mater 33(2):102–115
Rae PJ, Dickson PM (2020) Some observations about the drop-weight explosive sensitivity test. J Dyn Behav Mater
Kamlet MJ The relationship of impact sensitivity with structure of organic high explosives. 1. Polynitroaliphatic explosives. In: 6th International Symposium on Detonation, San Diego, California, 1976. ONR Report ACR 221, p 312
Kamlet MJ, Adolph HG (1979) Relationship of impact sensitivity with structure of organic high explosives: . polynitroaromatic explosives. Propellants and Explosives 4(2):30–34
Mathieu D (2016) Physics-based modeling of chemical hazards in a regulatory framework: comparison with quantitative structure-property relationship (QSPR) methods for impact sensitivities. Ind Eng Chem Res 55(27):7569–7577
Zhang CY (2009) Review of the establishment of nitro group charge method and its applications. J Hazard Mater 161(1):21–28
Murray JS, Concha MC, Politzer P (2009) Links between surface electrostatic potentials of energetic molecules, impact sensitivities and C-NO2/N-NO2 bond dissociation energies. Mol Phys 107(1):89–97
Yan QL, Zeman S (2013) Theoretical evaluation of sensitivity and thermal stability for high explosives based on quantum chemistry methods: a brief review. Int J Quantum Chem 113(8):1049–1061
Li G, Zhang C (2020) Review of the molecular and crystal correlations on sensitivities of energetic materials. J Hazard Mater 398:122910
Owens FJ, Jayasuriya K, Abrahmsen L, Politzer P (1985) Computational analysis of some properties associated with the nitro groups in polynitroaromatic molecules. Chem Phys Lett 116(5):434–438
Politzer P, Murray JS (1996) Relationships between dissociation energies and electrostatic potentials of C-NO2 bonds: applications to impact sensitivities. J Mol Struct 376:419–424
Murray JS, Lane P, Politzer P (1998) Effects of strongly electron-attracting components on molecular surface electrostatic potentials: application to predicting impact sensitivities of energetic molecules. Mol Phys 93(2):187–194
Ren FD, Cao DL, Shi WJ, Gao HF (2016) A theoretical prediction of the relationships between the impact sensitivity and electrostatic potential in strained cyclic explosive and application to H-bonded complex of nitrocyclohydrocarbon. J Mol Model 22(4):8
Politzer P, Murray JS, Serninario JM, Lane P, Grice ME, Concha MC (2001) Computational characterization of energetic materials. Theochem-J Mol Struct 573:1–10
Rice BM, Sahu S, Owens FJ (2002) Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules. Theochem-J Mol Struct 583:69–72
Li JS (2010) Relationships for the impact sensitivities of energetic C-nitro compounds based on bond dissociation energy. J Phys Chem B 114(6):2198–2202
Li J (2010) A quantitative relationship for the shock sensitivities of energetic compounds based on X–NO2 (X=C, N, O) bond dissociation energy. J Hazard Mater 180(1):768–772
Song XS, Cheng XL, Yang XD, He B (2006) Relationship between the bond dissociation energies and impact sensitivities of some nitro-explosives. Propellants Explos Pyrotech 31(4):306–310
Mathieu D (2012) Theoretical shock sensitivity index for explosives. J Phys Chem A 116(7):1794–1800
Mathieu D (2017) Sensitivity of energetic materials: theoretical relationships to detonation performance and molecular structure. Ind Eng Chem Res 56(29):8191–8201
Keshavarz MH (2010) Simple relationship for predicting impact sensitivity of nitroaromatics, nitramines, and nitroaliphatics. Propellants Explos Pyrotech 35(2):175–181
Keshavarz MH (2013) A new general correlation for predicting impact sensitivity of energetic compounds. Propellants Explos Pyrotech 38(6):754–760
Keshavarz MH, Ghaffarzadeh M, Omidkhah MR, Farhadi K (2017) New correlation between electric spark and impact sensitivities of nitramine energetic compounds for assessment of their safety. Z Anorg Allg Chem 643(19):1227–1231
Keshavarz MH, Ghaffarzadeh M, Omidkhah MR, Farhadi K (2017) Correlation between shock sensitivity of nitramine energetic compounds based on small-scale gap test and their electric spark sensitivity. Z Anorg Allg Chem 643(24):2158–2162
Keshavarz MH, Abadi YH (2018) Novel organic compounds containing nitramine groups suitable as high-energy cyclic nitramine compounds. ChemistrySelect 3(28):8238–8244
Shoaf AL, Bayse CA (2018) Trigger bond analysis of nitroaromatic energetic materials using wiberg bond indices. J Comput Chem 39(19):1236–1248
Stone AJ (1981) Distributed multipole analysis, or how to describe a molecular charge-distribution. Chem Phys Lett 83(2):233–239
Stone AJ, Alderton M (1985) Distributed multipole analysis - methods and applications. Mol Phys 56(5):1047–1064
Stone AJ (2000) The theory of intermolecular forces. International Series of Monographs on Chemistry. Oxford University Press, Oxford
Stone AJ (2005) Distributed multipole analysis: stability for large basis sets. J Chem Theory Comput 1(6):1128–1132
Borges I (2008) Conformations and charge distributions of diazocyclopropanes. Int J Quantum Chem 108(13):2615–2622
Anders G, Borges I (2011) Topological analysis of the molecular charge density and impact sensitivy models of energetic molecules. J Phys Chem A 115(32):9055–9068
Oliveira RSS, Borges Jr I (2020) Correlation between molecular charge properties and impact sensitivity of explosives: nitrobenzene derivatives. Propellants, Explosives, Pyrotechnics (at press)
Moraes TF, Borges I (2011) Nuclear Fukui functions and the deformed atoms in molecules representation of the electron density: application to gas-phase RDX (hexahydro-1,3,5-trinitro-1,3,5- triazine) electronic structure and decomposition. Int J Quantum Chem 111 (7–8):1444–1452
Giannerini T, Borges I (2015) Molecular electronic topology and fragmentation onset via charge partition methods and nuclear Fukui functions: 1,1-diamino-2,2-dinitroethylene. J Braz Chem Soc 26(5):851–859
Oliveira MAS, Borges I (2019) On the molecular origin of the sensitivity to impact of cyclic nitramines. Int J Quantum Chem 119(8):14
de Oliveira RSS, Borges I (2019) Correlation between molecular charge densities and sensitivity of nitrogen-rich heterocyclic nitroazole derivative explosives. J Mol Model 25(10):314
Borges I, Silva AM, Aguiar AP, Borges LEP, Santos JCA, Dias MHC (2007) Density functional theory molecular simulation of thiophene adsorption on MoS2 including microwave effects. Theochem-J Mol Struct 822(1–3):80–88
Borges I, Silva AM (2012) Probing topological electronic effects in catalysis: thiophene adsorption on NiMoS and CoMoS clusters. J Braz Chem Soc 23(10):1789–1799
Borges I, Silva AM, Modesto-Costa L (2018) Microwave effects on NiMoS and CoMoS single-sheet catalysts. J Mol Model 24(6):8
Silva AM, Borges I (2011) How to find an optimum cluster size through topological site properties: Mosx model clusters. J Comput Chem 32(10):2186–2194
Dlott DD (2003) Fast molecular processes in energetic materials. In: Politzer P, Murray JS (eds) energetic materials. Part 2. Detonation, combustion, vol 12. Elsevier, Amsterdam,
Brill TB, James KJ (1993) Thermal decomposition of energetic material. 61. Perfidy in the amini-2,4,6-trinitrobenzene series of explosives. J Phys Chem 97(34):8752–8758
Pro S (1999). Wavefunction, Irvine
Hohenberg P, Kohn W (1964) Inhomogeneous electron gas. Phys Rev B 136 (3B):B864-&
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140(4A):1133–1138
Becke AD (1993) A new mixing of Hartree-Fock and local density-functional theories. J Chem Phys 98(2):1372–1377
Frisch MJ (et al., Gaussian 03, Revision B.01, 2003) Gaussian 03, Revision B.01
Price SL, Stone AJ (1983) A distributed multipole analysis of charge-densities of the azabenzene molecules. Chem Phys Lett 98(5):419–423
Atkins PW, dePaula J (2006) Physical chemistry8th edn. W. H, Freeman and Company, New York (NY)
Politzer P, Murray JS, Lane P, Sjoberg P, Adolph HG (1991) Shock-sensitivity relationships for nitramines and nitroaliphatics. Chem Phys Lett 181(1):78–82
Murray JS, Politzer P (2011) The electrostatic potential: an overview. Wiley Interdiscip Rev-Comput Mol Sci 1(2):153–163
Clark T, Murray JS, Politzer P (2018) A perspective on quantum mechanics and chemical concepts in describing noncovalent interactions. Phys Chem Chem Phys 20(48):30076–30082
Politzer P, Murray JS, Clark T, Resnati G (2017) The sigma-hole revisited. Phys Chem Chem Phys 19(48):32166–32178
Politzer P, Murray JS (2018) The Hellmann-Feynman theorem: a perspective. J Mol Model 24(9):7
Xiang D, Zhu WH (2017) Thermal decomposition of isolated and crystal 4,10-dinitro-2,6,8,12-tetraoxa-4,10-diazaisowurtzitane according to ab initio molecular dynamics simulations. RSC Adv 7(14):8347–8356
Storm CB, Stine JR, Kramer JF (1990) In: chemistry and physics of energetic materials. Kluwer Acad. Publ, Dordrecht, pp 605–640
Fischer JW, Hollins RA, LoweMa CK, Nissan RA, Chapman RD (1996) Synthesis and characterization of 1,2,3,4-cyclobutanetetranitramine derivatives. J Organomet Chem 61(26):9340–9343
Yau AD, Byrd EFC, Rice BM (2009) An investigation of KS-DFT electron densities used in atoms-in-molecules studies of energetic molecules. J Phys Chem A 113(21):6166–6171
Dubovitskii FI, Korsunskii BL (1981) Kinetics of the thermal decomposition ofN-nitro-compounds. Russ Chem Rev 50(10):958–978
Astakhov AM, Dyugaev KP, Kuzubov AA, Nasluzov VA, Vasiliev AD, Buka ÉS (2009) Theoretical studies of the structure of nitrimines. I. Structure of 2-nitroguanidine and its alkyl derivatives. J Struct Chem 50(2):201–211
Bracuti AJ (1999) Crystal structure refinement of nitroguanidine. J Chem Crystallogr 29(6):671–676
Moxnes JF, Froyland O, Risdal T (2017) A computational study of ANTA and NTO derivatives. J Mol Model 23(8):8
Schmidt NB, Lee GS, Mitchell AR, Gilardi R (2001) Synthesis and properties, of a new explosive, 4-amino-3,5-d i n it ro-1 H-Pyrazole(LLM-116). Lawrence Livermore National Laboratory, Lawrence Livermore National Laboratory
Bulusu NB (1990) Chemistry and physics of energetic materials, vol 309. 1st edn. Kluwer Academic Publishers, Dordrecht
Depaz JLG, Ciller J (1994) Structure and tautomerismo of anta (aminonitrotriazole). Propellants Explos Pyrotech 19(1):32–41
Su XF, Cheng XL, Ge SH (2009) Theoretical investigation on structure and properties of 2,4,5-trinitroimidazole and its three derivatives. Theochem-J Mol Struct 895(1–3):44–51
Damavarapu R, Surapaneni CR, Duddu RG, Zhang M, Dave PR (2007) Melt-cast explosive material. United States Patent,
Kalescky R, Kraka E, Cremer D (2013) Local vibrational modes of the formic acid dimer – the strength of the double hydrogen bond. Mol Phys 111(9–11):1497–1510
Tao Y, Zou W, Kraka E (2017) Strengthening of hydrogen bonding with the push-pull effect. Chem Phys Lett 685:251–258
Freindorf M, Kraka E, Cremer D (2012) A comprehensive analysis of hydrogen bond interactions based on local vibrational modes. Int J Quantum Chem 112(19):3174–3187
Politzer P, Seminario JM, Bolduc PR (1989) A proposed interpretation of the destabilizing effect of hydroxyl-groups of nitroaromatic molecules. Chem Phys Lett 158(5):463–469
Murray JS, Lane P, Politzer P, Bolduc PR (1990) A relationship between impact sensitivy and the electrostatic potentials at the midpoints of C-NO2 bonds in nitroaromatics. Chem Phys Lett 168(2):135–139
Zhang CY, Shu YJ, Huang YG, Zhao XD, Dong HS (2005) Investigation of correlation between impact sensitivities and nitro group charges in nitro compounds. J Phys Chem B 109(18):8978–8982
Acknowledgments
We thank an anonymous referee of another publication [68] for suggesting to pursue further a Coulombic force for modeling the sensitivity of explosives. The support of the Brazilian Army to this work through our Institute is greatly acknowledged.
Funding
Support for this research is from the Brazilian agency CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) through research grants 304148/2018-0 and 409447/2018-8. R. S. S. O. has a Ph.D. scholarship from Capes (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior).
Author information
Authors and Affiliations
Contributions
All authors contributed equally to the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Code availability
The Gaussian, Sparta, and the GDMA2 (http://www-stone.ch.cam.ac.uk/pub/gdma/) programs were used to generate the data.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
The online version of this article has additional Supplementary Material that includes acronyms of the molecules; monopole values and Coulombic force values of the N-NO2 and C-NO2 bonds; normalized Coulombic force values for the analyzed molecules and converged geometry data for the nitramine and nitroazole molecules. (DOCX 4167 kb)
Rights and permissions
About this article

Cite this article
Oliveira, M.A.S., Oliveira, R.S.S. & Borges, I. Quantifying bond strengths via a Coulombic force model: application to the impact sensitivity of nitrobenzene, nitrogen-rich nitroazole, and non-aromatic nitramine molecules. J Mol Model 27, 69 (2021). https://doi.org/10.1007/s00894-021-04669-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-021-04669-5