
Abstract
This study aims to experimentally and theoretically examine the nature and energy of intermolecular bond interactions between thiourea and water molecules using natural bond orbital (NBO), non-linear optical (NLO), atoms in molecules (AIM), and reduced density gradient (RDG) analyses based on the quantum chemical approach and spectroscopic analysis on X-ray and FTIR. Geometry optimizations of Thio-(H2O)1–5 complexes were carried out in the gas phase by B3LYP/6-311++G(d,p) level of density functional theory. The nature of the molecular interactions between the water and thiourea through hydrogen bonding has been investigated using RDG and AIM methods. NBO analysis shows that the Thio-(H2O)5 complex has higher stabilization energy values than the other complexes. The non-linear optical properties, such as dipole moment (μ), the polarizability (α0), and the first hyperpolarizability (βtot), and thermodynamic functions, such as entropy (S), specific heat capacity (Cv), and thermal energy (E), were calculated using the same method. It was observed that thermodynamic parameters, polarizability, and the first hyperpolarizability increased with the number of water molecules. X-ray diffraction analysis confirmed that thiourea is single crystal, and the thiourea/water complexes are crystalline in nature. Besides, the infrared spectrum shows the existence of water molecules and it is used to get details of the structure of the complex.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Thiourea (thiocarboxylic acid diamide) is an important substance in organic synthesis and is intensively used in the processes of obtaining drugs. It is a fairly simple organic molecule (with a thioamide group); due to its active use in organic synthesis, its structure and properties have been actively studied by various methods for many years [1,2,3,4]. Due to the properties of its complex, thiourea is also actively used on an industrial scale [5]. It, like its derivatives, has been widely used as corrosion inhibitors [6] in industrial equipment for disposal of scale [7]. It is known that thiourea derivatives have a number of unique properties for use in agriculture, analytical chemistry, rubber industry, in the development of photographs [5], as well as in pharmacology [8,9,10,11,12,13,14,15]. Studies of thiourea complexes with various types of molecules are given special attention [16, 17]. Much attention in such studies is given to interactions occurring at the intermolecular level, in particular, hydrogen bonds, since this characteristic is important for a number of fundamental areas, such as physical, chemical, and biological [18,19,20,21]. One of the fundamental and most important processes in chemistry are reactions in aqueous solution and solvation. Currently, these processes are actively studied, both experimentally and theoretically. A complete understanding of the various processes occurring in aqueous solution is currently lacking, despite intensive research in this area [22, 23]. However, quantum mechanical calculations give the most complete ideas about the mechanisms of these processes, because it allows the study of such mechanisms at the molecular level [18]. Thus, it was previously shown [24,25,26] that the addition of water molecules can increase the size of the clusters of thiourea/water complexes. Also, using the density functional theory, the relative stability of thiourea in water was studied using the example of thiourea/water clusters. It was shown that this thiourea/water complex is gradually stabilized by the addition of water molecules, as evidenced by an increase in the binding energy [18]. In the literature, numerous theoretical studies have been conducted on the complex (even thiourea/water complex [18]) and applications. However, we have brought different perspectives to these complexes by using different methods such as natural bond orbital (NBO), AIM, X-ray, and RDG in here. The aim of this study is to investigate thiourea/water complexes by means of X-ray diffraction and intermolecular hydrogen bond interaction with NBO, AIM, and RDG analyses.
Experimental details
Samples of the thiourea/water complex were obtained by dissolving thiourea in distilled water at 50 °C, followed by precipitation of the complex at room temperature. The introduction of water into thiourea crystals is proved by the X-ray diffraction method. The X-ray diffraction (XRD) phase analysis was performed on a DRON-3 X-ray diffractometer using CuKα monochromatized radiation (λ = 0.154 nm), voltage 30 kV, and current 25 mA. The scanning step is 0.02°; intervals for 1 s per data point. The measurement was carried out in the interval of the Bragg angles 2Θ from 5.00 to 70.00°. The FTIR spectra of pure thiourea and thiourea/water complex were registered using a Shimadzu IR Tracer-100 spectrometer (Japan) within the wavelength range of 400–4000 cm−1. The spectral data was analyzed using the OPUS program (version 5.0). Solid samples for analysis were prepared in the form of pills in a KBr matrix (2 mg sample/1000 mg KBr).
Theoretical calculations
All density functional theory (DFT) computations were made using Gaussian 09 program package [27] and GaussView molecular imaging program [28]. To calculate optimized geometrical molecular structure, DFT with Becke’s three-parameter hybrid exchange function combined with the Lee-Yang-Parr correlation functional (B3LYP) [29, 30] and 6-311++G(d,p)-extended basis set were utilized. The fact that the vibration frequencies obtained by frequency calculations are all positive indicates that all fixed points are present on the potential energy surface as true minima. In such complex, to determine the energy of intermolecular interaction, many analyses have been used such as NBO analysis, by computing donor acceptor interaction energy and the atoms in molecules (AIM) theory of Bader. Using this theory, many topological parameters were calculated by the means of the Multiwfn program [31]. The non-covalent interactions and strong repulsions in the complexes are determined using reduced density gradient (RDG) analysis. Finally, the results were drawn using VMD program [32, 33].
Results and discussion
Determination of most stable complexes of Thio-(H2O)n
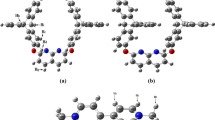
In order to investigate the interactions of the thiourea molecule in water, complexes of thiourea/water were designed using the sites of the thiourea molecule that have the potential to make hydrogen bonds with water, and the most stable complexes were identified. The most stable complexes possible, such as Thio-(H2O)1, Thio-(H2O)2, Thio-(H2O)3, Thio-(H2O)4, and Thio-(H2O)5, were optimized at B3LYP level of theory with the basis set 6-311++G(d,p) and are represented by atomic numbers in Fig. 1a–e. Optimized parameters (bond lengths, bond angles) for the pure thiourea and Thio-(H2O)1–5 were obtained using the same method and compared to each other. The optimized bond parameters of the pure thiourea and Thio-(H2O)1–5 molecules are given in Table 1. As seen in the Table 1, the pure thiourea and Thio-(H2O)1–5 complexes have two C-N, one C=S, and four N-H bond lengths. The bond lengths of C-N1 and C-N2 in pure thiourea are slightly longer than those in the complexes, and these bonds are gradually shortened in complexes with an increasing number of water molecules. The bond length of C=S in pure thiourea is found to be as 1.67 Å and its value slightly lower than that of complexes, because it appears to be in a more sensitive interaction against hydration. The highest and lowest bond lengths were calculated for N2-H in Thio-(H2O)5 found to be 2.099 Å and for N1-H and N2-H in pure thiourea and N2-H in Thio-(H2O) found to be 1.007 Å, respectively. The highest and lowest bond angles that were calculated for N1-C-S in Thio-(H2O) were found to be 123.432°, and for N1-C-N2 in pure thiourea, they were found to be 114.686°, respectively. Thio-(H2O)1–5 complexes that were formed through intermolecular hydrogen-bonding interactions of the water molecules with sulfur and hydrogen atoms of thiourea were computed at the B3LYP/6-311++G(d,p) level of theory and are listed in Table 2. From Fig. 1 and Table 2, Thio-(H2O)1 complex has two intramolecular hydrogen bonds O-H⋯S and N-H⋯S of lengths 2.369 and 1.937 Å, respectively. Thio-(H2O)2–5 complexes have four, five, nine, and eight intramolecular hydrogen bonds, respectively. The largest intramolecular hydrogen bond N5-H⋯O11 is formed in Thio-(H2O)4 complex with bond length of 3.519 Å. Geometric parameters that define interactions between thiourea and water molecules are the distance and angle between donor and acceptor groups in the aqueous solutions of thiourea. Geometric parameters determine the shape of the molecule, and it is seen that all complexes form a ring structure. The intermolecular hydrogen-bonding interaction energies calculated by the B3LYP/6-311++G(d,p) method and Table 3 gives the intermolecular hydrogen-bonding interaction energy in Thio-(H2O)1–5 complexes calculated using the formula below:

Optimized structures of thiourea/water complexes. a Thiourea-(H2O). b Thiourea-(H2O)2. c Thiourea-(H2O)3. d Thiourea-(H2O)4. e Thiourea-(H2O)5
From Table 3, the interaction energy of the ring dimer complex is − 38.94 kcal mol−1. The interaction energy of the ring dimer complex (Fig. 1a) is lower than that of the other ring complexes. In this ring dimer, two and N-H⋯O bonds cause the formation of a ring shape between the water and thiourea molecules. The O-H⋯S hydrogen bond is 2.369 Å and larger than the O-H⋯O hydrogen bond. In the Thio-(H2O)2 (Fig. 1b), the interaction energy is − 76.75 kcal mol−1 and its ring structure formed by four hydrogen bonds such as N-H⋯O8, N-H⋯O11, O8-H⋯S, and O11-H⋯S. The bond lengths of N-H⋯O8 and N-H⋯O11 are equal to 1.937 Å; likewise, O8-H⋯S and O11-H⋯S equal to 2.391 Å. In the Thio-(H2O)3 (Fig. 1c), the interaction energy is − 119.12 kcal mol−1 and its ring structure formed by five hydrogen bonds such as N-H⋯O11, N-H⋯O14, O8-H⋯S, O14-H⋯S, and O8-H⋯O11. In the Thio-(H2O)4 (Fig. 1d), the interaction energy is − 176.38 kcal mol−1 and its ring structure formed by nine hydrogen bonds such as N2-H⋯O14, N5-H⋯O14, N5-H⋯O17, N5-H⋯O8, N5-H⋯O11, O8-H⋯S, O11-H⋯O8, O17-H⋯O17, and O14-H⋯O. In the Thio-(H2O)5 (Fig. 1e), the interaction energy is − 192.59 kcal mol−1 and its ring structure formed by eight hydrogen bonds such as N2-H⋯O17, N2-H⋯O14, N5-H⋯O14, N5-H⋯O11, O8-H⋯S, O20-H⋯S, O11-H⋯O8, and O17-H⋯O20.
X-ray and FTIR analyses
X-ray diffraction analysis showed that thiourea single crystals and the thiourea/water complex are crystalline in nature; their diffraction peaks were indexed and are shown in Fig. 2. As shown in Fig. 2, the introduction of water molecules into the crystalline structure of thiourea is affected by a change in the intensity of peaks in X-ray diffractograms: an increase intensity in peaks at 4.27, 3.494, 3.14, and 3.09° (2 theta) and a decrease intensity in peaks at 3.83 and 2.474° (2 theta); also, there is a slight peak shift from 2.856 to 2.847 and from 2.32 to 2.314° (2 theta). Clearly defined, sharp peaks in X-ray diffraction patterns indicate the single-phase and good crystalline nature of pure thiourea crystals [34, 35] and the thiourea/water complex. An analysis of the FTIR spectra of thiourea and the thiourea/water complex using known data on the characteristic frequencies of individual functional groups made it possible to assign absorption bands and establish some patterns [36, 37]. The FTIR spectra (Fig. 3; Table 4), which contain absorption bands at 3380, 3276, 1614, and 730 cm−1 for thiourea and thiourea/water, correspond to asymmetric and symmetric stretching vibrations of NH2; absorption bands at 1469 and 1412 cm−1 correspond to vibrations of C-N groups; 489 cm−1 corresponds to vibrations of N-C-S groups; and 1082 cm−1, corresponds to stretching vibrations of C-N bonds. In the FTIR spectrum of thiourea/water, an increase in the intensity of vibration of bands is observed at 3380, 3276, 1614, and 1082 cm–1 as a result of superposition of the vibration of OH groups [37].

XRD analysis of thiourea (a) and thiourea/water complex (b)

FTIR spectra: thiourea (1) and thiourea/water complex (2)
NBO analysis
Natural bond orbital analysis is an important method that accurately demonstrates interaction between bonds and intra-/intermolecular hydrogen bonding and allows to investigate the delocalization of electron density for takes place between the lone pair bond orbital or the occupied and the unoccupied antibonding orbital [38]. These interactions can be quantitatively identified in the way of NBO analysis denoted by the second-order perturbation stabilization energy (E(2)). The stabilization energies (E(2)) correlated with delocalization can be given as follows:
where F(i, j) → the off-diagonal NBO Fock matrix element, εj and εi → the diagonal elements, and qi → donor orbital occupancy. The large second-order perturbation stabilization energy (E(2)) values obtained by NBO analysis indicate greater interaction between electron-acceptors and electron-donors, and the degree of conjugation of the entire system is higher.
Natural bond orbital (NBO) analysis of Thio-(H2O)1–5 complexes was performed at using NBO 3.1 program [39] as applied in the Gaussian 09 program at the B3LYP/6-311++G(d,p) method of theory owing to understand second-order interactions of the H-bonded complexes in thiourea/water mixture, and the possible intensive interaction is listed in NBO in Table 5. It has been found that hydrogen bonds such as N-H⋯O and O-H⋯S between thiourea and water molecules and hydrogen bond between water molecules are formed, and these bonds help the stability of the possible complexes. Looking at the NBO analysis of Thio-(H2O)1 complex, it is noted the strong intermolecular hydrogen-bonding interactions between LP(O8) lone pair as donor orbital and σ*(N2-H4) antibonding orbital as acceptor, which have the highest E(2) value around 9.71 kcal mol−1 and between LP(S11) lone pair as donor orbital and σ*(O8-H9) antibonding as acceptor with a stabilization energy of 8.99 kcal mol−1. In Thio-(H2O)2 complex, it is noted the strong intermolecular hydrogen-bonding interactions between LP(O8) and LP(O11) lone pair as donor orbitals and σ*(N2-H4) and σ*(N5-H6) antibonding orbitals as acceptor, which have the highest E(2) values around 9.87 kcal mol−1 and, that is, they are equal to each other. The intermolecular hydrogen-bonding interactions between LP(S14) lone pair as donor orbitals and σ*(O8-H10) and σ*(O11-H12) antibonding orbitals as acceptor are quite high with 7.7 kcal mol−1 energy values and equal to each. Besides, it is noted the interactions between LP(N2) and LP(N5) lone pair as donor orbitals and σ*(C1-S14) antibonding orbitals as acceptor, which have the highest E(2) values around 80.77 kcal mol−1 and, that is, they are equal to each other. In Thio-(H2O)3 complex, it is noted the strong intermolecular hydrogen-bonding interactions between LP(O8) lone pair as donor orbital and σ*(O11-H12) antibonding orbital as acceptor, which have the highest E(2) values around 14.39 kcal mol−1. Besides, the interactions between LP(N5) lone pair as donor orbitals and σ*(C1-S17) antibonding orbitals as acceptor are quite high with 74.19 kcal mol−1 energy value. In Thio-(H2O)4 complex, it is noted the strong intermolecular hydrogen-bonding interactions between LP(S20) lone pair as donor orbital and σ*(O8-H10) antibonding orbital as acceptor, which have the highest E(2) values around 13.36 kcal mol−1. The strong interactions between π*(C1-S20) bonding orbital as donor orbital and σ*(C1-S20) antibonding orbital as acceptor, which have the highest E(2) values around 66.18 kcal mol−1. Besides, the interactions between LP(N5) lone pair as donor orbitals and π *(C1-S20) antibonding orbitals as acceptor are quite high with 29.22 kcal mol−1 energy value. In Thio-(H2O)5 complex, it is noted the strong intermolecular hydrogen-bonding interactions between LP(O8) and LP(O20) lone pairs as donor orbital and σ*(O11-H13) and σ*(O17-H18) antibonding orbital as acceptor, which have the highest E(2) values around 14.24 kcal mol−1. It is noted the strong interactions between LP(N2) lone pair as donor orbital and σ*(C1-N5) antibonding orbital as acceptor, which have the highest E(2) values around 82.21 kcal mol−1. Besides, the interactions between LP(S23) lone pair as donor orbitals and σ*(C1-N5) antibonding orbitals as acceptor are quite high with 54.79 kcal mol−1 energy value. As seen in the NBO analysis, the highest energy interactions were seen in the Thio-(H2O)5 complex.
AIM and RDG topological analysis
The theory of atoms in molecules (AIM) recommended by Bader has been widely used to determine the type of different interactions in several molecular systems and to analyze the bonding interactions from the point of the real space functions as electron density at the bond critical points (BCPs) [40]. The properties of hydrogen bonds between compounds can be seen using topological parameters such as Laplacian of electron density ∇2ρ(r), the electron density ρ(r), Lagrangian kinetic energy G(r), the potential energy density V(r), Hamiltonian kinetic energy H(r) = G(r) + V(r), and bond energy Eint = V(r)/2. For AIM analyses, the optimized thiourea and water complexes were used to obtain at the intramolecular BCPs and ring critical points (RCPs) between thiourea and water molecules. The calculation of AIM analysis was done using the Multiwfn program. The molecular graph of the Thio-(H2O)1–5 complexes in the RCP and BCP of the noted molecular interactions obtained with the Multiwfn program is illustrated in Fig. 4, and the topological parameters of these complexes in all the BCP and RCPs are listed in Table 6.

AIM molecular graphs of thiourea/water complexes (RCP is shown as small yellow spheres, BCP is shown as small red spheres, bond paths is shown as yellow lines). a Thiourea-(H2O). b Thiourea-(H2O)2. c Thiourea-(H2O)3. d Thiourea-(H2O)4. e Thiourea-(H2O)5
According to Rozas et al. [41], the hydrogen bond interactions can be classified as follows:
- (1)
Weak hydrogen bonds are determined by ∇2ρ(r) > 0 and H(r) >0
- (2)
Moderate hydrogen bonds are determined by ∇2ρ(r) > 0 and H(r) < 0
- (3)
Strong hydrogen bonds are determined by ∇2ρ(r) < 0 and H(r) < 0
Electron density ρ(r) and its Laplacian ∇2ρ(r) help determine the nature of interactions. In general, the large values of electron density ρ(r) and its Laplacian ∇2ρ(r) show the power of hydrogen interactions [42]. The positive values of Laplacian ∇2ρ(r) are ascribed to the reducing of the charge in the internuclear region, while the negative values are indicative of a strong covalent character. In the Thio-(H2O) complex, we observed N2-H4⋯O8 and O2-H9⋯S11 type of interaction, where the electron density values are 0.0201 and 0.0144 a.u. and the values of Laplacian are 0.1220 and 0.0764 a.u., respectively. In the Thio-(H2O)2 complex, we observed two N-H⋯O (N2-H4⋯O8 and N5-H6⋯O11) and two O-H⋯S (O8-H10⋯S14 and O11-H12⋯S14) types of interaction, where the electron density values are equal to 0.0200 and 0.0143 a.u. and the values of Laplacian are equal to0.1219 and 0.0736 a.u., respectively. In the Thio-(H2O)3 complex, we observed two N-H⋯O (N5-H6⋯O14 and N2-H4⋯O11), two O-H⋯S (O14-H15⋯S17 and O8-H10⋯S17), and O11-H12⋯O8 types of interaction, where the electron density values are 0.0198 and 0.0223 a.u., 0.0146 and 0.0150 a.u., and 0.0256 a.u. and the values of Laplacian are 0.1208 and 0.1456 a.u., 0.0757 and 0.0866 a.u., and 0.1742 a.u., respectively. In the Thio-(H2O)4 complex, we observed three N-H⋯O (N2-H3⋯O14, N5-H7⋯O4, and N5-H6⋯O8), O8-H10⋯S20, and three O-H⋯O (O14-H16⋯O17, O17-H18⋯O11, and O11-H12⋯O14) types of interaction, where the electron density values are 0.0160, 0.0183, 0.0107 a.u.; 0.0163 a.u.; and 0.0270, 0.0259, 0.0241 a.u. and the values of Laplacian are 0.0810, 0.0975, 0.0478 a.u.; 0.1012 a.u.; and 0.1962, 0.1856, 0.1644 a.u., respectively. In the Thio-(H2O)5 complex, we observed four N-H⋯O (N5-H7⋯O14, N2-H3⋯O14, N5-H6⋯O11, and N2-H4⋯O17), two O-H⋯S (O8-H9⋯S23 and O20-H22⋯S23), and two O-H⋯O (O11-H13⋯O8 and O17-H18⋯O20) types of interaction, where the electron density values are equal to 0.0139 and 0.0207 a.u.; equal to 0.0158 a.u.; and equal to 0.0255 a.u. and the values of Laplacian are 0.0670, 0.0671, 0.1282, 0.1285 a.u.; 0.0945, 0.0944 a.u.; and 0.1722, 0.1723 a.u., respectively. As known, positive values of Laplacian ∇2ρ(r) and low values of electron density ρ(r) in BCPs indicate the presence of hydrogen bond interactions [40]. According to the BCP analysis of Table 6, all hydrogen bond interactions in all the Thio-(H2O)1–5 complexes are with bond energies in a range of − 11.66 to − 53.72 kJ mol−1. The two O-H⋯O (O14-H16⋯O17 and O17-H18⋯O11) types of interaction in Thio-(H2O)4 complex are considered the strongest interactions with hydrogen bond energies − 53.72 and − 50.19 kJ mol−1, respectively. The powerfulness of these hydrogen bonds in Thio-(H2O)4 complex is verified by the highest positive values of the Laplacian. The non-covalent interaction index (NCI) is used to characterize intermolecular interactions and evaluate the nature of weak interactions. The NCI index supplies more evidence of non-covalent interaction, and it is based upon the reduced density gradient (RDG). The reduced density gradient (RDG) is a basic non-dimensional quantity, which consists of the density and first derivative, and it is expressed by the formula below [43].
The electron density quantity of the RDG versus sign (λ2) ρ peaks gives us information about the nature and power of interactions of molecules. The interaction of the power in the molecular system, which indicates the stronger attractiveness of blue color and the push of red, is analyzed with Multiwfn and VMD software. The value of sign (λ2) ρ is very important in predicting the nature of interaction, that is, if the sign (λ2) ρ > 0 shows a repulsive interaction (non-bonded) and the sign (λ2) ρ < 0 shows an attractive interaction (bonded). The RDG scatter graphs of Thio-(H2O) complexes are shown in Fig. 4. As seen on the left side of Fig. 5, the blue colors indicate the hydrogen-bonding interaction, green colors are van der Waals interactions, and the red color is defined as strong repulsion (steric effect). As seen on the right side of Fig. 5, the blue color shows the hydrogen bond, while the green color corresponds to the van der Waals interaction and the red color the steric cyclic effect. When looking at complexes, van der Waals interactions were more seen between the thiourea and water molecules. The results show the attractive van der Waals and repulsive interactions in our complexes.


Reduced density gradient analysis (RDG) to know weak and strong interaction in these complexes
Thermodynamic and NLO properties
The standard thermodynamic functions such as entropy (S), specific heat capacity (Cv), thermal energy (E), and the other parameters were calculated by using B3LYP methods with the 6-311++G(d,p) basis set and are listed in Table 7. These functions are important in predicting the reactivity of chemical reactions and determining the probability of different reaction routes [44]. As seen in Table 7, the values of Cv, S, and E all increase with the increase in the number of water molecules from 1 to 5, which is attributed to the increase in molecular vibration as the number of water molecules increases. The variation of thermodynamic parameters as a function of the number of water molecules are shown in Fig. 6. Besides, the non-linear optical (NLO) properties such as dipole moment (μ) the polarizability (α0) and the first hyperpolarizability (βtot) for the thiourea/water complexes were calculated by using the mentioned method and given in Table 8. The variation of the polarizability (α0) and the first hyperpolarizability (βtot) as a function of the number of water molecules are shown in Fig. 7.

The variation of thermodynamic parameters as a function of the number of water molecules

The variation of the first hyperpolarizability (βtot) and the polarizability (α0) as a function of the number of water molecules
According to Fig. 7 and Table 8, the values of the polarizability (α0) and the first hyperpolarizability (βtot) increase with the increase in the number of water molecules from 1 to 5.
Conclusions
The interactions of the thiourea molecule in water investigated using NBO, AIM, and RDG analyses based on DFT method. All calculations of Thio-(H2O)1–5 complexes were performed in the gas phase by B3LYP/6-311++G(d,p) level of theory. Firstly, the most stable complexes possible with water were identified, and the shape of the thiourea/water complexes forming a ring structure was determined with the help of geometric parameters. The structures of these complexes were experimentally illuminated by FTIR and X-ray analyses. NBO analysis showed the presence of strong hydrogen bonds in the aqueous solutions of thiourea. Besides, thiourea/water complex is gradually stabilized by the addition of water molecules, as evidenced by an increase in the binding energy. The interactions formed within the complexes such as Van der Walls, hydrogen bond interactions, and steric effect are classified by the means of RDG surface analysis. The strongest and weakest intermolecular hydrogen bond interactions were observed in Thio-(H2O)4 complex with 53.72 and 11.66 kJ mol−1, respectively. The first hyperpolarizability values of Thio-(H2O)1–5 complexes are 1.04, 1.33, 1.97, 2.50 and 2.72 × 10−30 e.s.u. which is larger than the value of NLO material urea (0.13 × 10−30 e.s.u.)
References
Olah GA, Burrichter A, Rasul G, Christe KO, Prakash GS (1997) Preparation, NMR, Raman, and DFT/IGLO/GIAO-MP2 study of mono-and diprotonated thiourea and theoretical investigation of triprotonated thiourea1. J Am Chem Soc 119(19):4345–4352
Hata M, Moribe K, Ando S, Tozuka Y, Yamamoto K (2014) Density functional theory study of equimolar complexation of urea or thiourea with 2-alcoxybenzamide. J Struct Chem 55(8):1506–1513
Martin ML, Filleux-Blanchard ML, Martin GJ, Webb GA (1980) Application of 15N spectroscopy and dynamic NMR to the study of ureas, thioureas and their Lewis acid adducts. Org Magn Reson 13(6):396–402
Ha TK, Puebla C (1994) A theoretical study of conformations and vibrational frequencies in (NH2) 2C = X compounds (X = O, S, and Se). Chem Phys 181(1–2):47–55
Sahu S, Rani Sahoo P, Patel S, Mishra BK (2011) Oxidation of thiourea and substituted thioureas: a review. J Sulfur Chem 32(2):171–197
Klern H, Lux J, Noll D, Reider W, Phillips H (1971) Proc. of the American Power Conference, Illinois Institute of Technology, Chicago, IL, April 20–22. 33:702–709
Knox JA, Smith JA, Stout RF (1974) US Patent №3,730,901. Chem Abstr 81:53183
Dawood KM (2019) Bis-thiourea derivatives and their utility in synthesis of mono-heterocyclic, bis-heterocyclic, and fused heterocyclic systems. J Heterocyclic Chem 56(6):1701–1721
Liav A, Angala SK, Brennan PJ, Jackson M (2008) ND-aldopentofuranosyl-N′-[p-(isoamyloxy) phenyl]-thiourea derivatives: potential anti-TB therapeutic agents. Bioorg Med Chem Lett 18(8):2649–2651
Tsogoeva SB, Hateley MJ, Yalalov DA, Meindl K, Weckbecker C, Huthmacher K (2005) Thiourea-based non-nucleoside inhibitors of HIV reverse transcriptase as bifunctional organocatalysts in the asymmetric Strecker synthesis. Bioorg Med Chem 13(19):5680–5685
Falzon D, Hill G, Pal SN, Suwankesawong W, Jaramillo E (2014) Pharmacovigilance and tuberculosis: applying the lessons of thioacetazone. Bull World Health Organ 92:918–919
Narimani H, Kohzadi H (2014) A practical and convenient method for the synthesis of anesthetic drug thiopental: using thiourea and sodium ethoxide. Iran Chem Commun 2:48–55
Živec M, Anzic B, Gobec S (2010) A novel scalable synthesis of pramipexole. Org Process Res Dev 14(5):1125–1129
Hassan AA, El-Sheref EM (2010) Chemistry and heterocyclization of dithiobiurea and thioureidoalkylthiourea. J Heterocyclic Chem 47(4):764–784
Sharma SK, Steinbergs N, Wu Y, Crowley ML, Hanson AS, Casero Jr RA, Woster PM (2010) (Bis)urea and (Bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators. J Med Chem 53(14):5197–5212
Jara P, Merchan J, Yutronic N, Gonzalez G (2000) Macroscopic evidence of inclusion phenomena in urea and thiourea matrices. J Incl Phenom Macrocycl Chem 36(1):99–100
Shakeel A, Altaf AA, Qureshi AM, Badshah A (2016) Thiourea derivatives in drug design and medicinal chemistry: a short review. J Drug Des Med Chem 2(1):10
Weiqun Z, Wen Y, Lihua Q (2005) Structure and stability of thiourea with water, DFT and MP2 calculations. J Mol Struct THEOCHEM 730(1–3):133–141
Pawlowski PM, Okimoto SR, Tao FM (2003) Structure and stability of sulfur trioxide–ammonia clusters with water: implications on atmospheric nucleation and condensation. J Phys Chem A 107(27):5327–5333
Ju XH, Xiao HM (2002) Ab initio study on interactions in difluoroamine clusters from one to four molecules. Propellants Explos Pyrotech 27(6):320–326
Xia QY, Xiao HM, Ju XH, Gong XD (2004) Density functional theory study of the structures and properties of (H2AlN3)n (n = 1–4) clusters. J Phys Chem A 108(14):2780–2786
Willard AP, Chandler D (2014) The molecular structure of the interface between water and a hydrophobic substrate is liquid-vapor like. J Chem Phys 141(18):18C519
Ghosh SR, Debnath B, Jana AD (2020) Water dimer isomers: interaction energies and electronic structure. J Mol Model 26(1):1–9
Hammami F, Ghalla H, Nasr S (2015) Intermolecular hydrogen bonds in urea–water complexes: DFT, NBO, and AIM analysis. Comput Theor Chem 1070:40–47
Kaur D, Khanna S (2014) Hydrogen bonding of formamide, urea, urea monoxide and their thio-analogs with water and homodimers. J Chem Sci 126(6):1815–1829
Ivanov EV, Abrosimov VK (2013) Apparent molar volumes and expansibilities of thiourea, 1,3-dimethylurea, and 1,3-dimethylthiourea in water at temperatures from T=(278.15 to 318.15) K and atmospheric pressure. J Chem Eng Data 58(5):1103–1111
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision C.01. Gaussian, Inc., Wallingford
GaussView, Guassian, Inc. (Carnergie Office Parck-Building6 Pittsburgh PA 151064 USA), Copyright © 2000-2003 Semichem. Inc.
Becke AD (1993) Becke’s three parameter hybrid method using the LYP correlation functional. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37(2):785
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33(5):580–592
Runge E, Gross EK (1984) Density-functional theory for time-dependent systems. Phys Rev Lett 52(12):997
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14(1):33–38
Jayalakshmi D, Kumar J (2006) Growth and characterization of bis thiourea zinc acetate (BTZA). J Cryst Res Technol 41(1):37–40
Ravi B, Jegatheesan A, Neelakandaprasad B, Sadeeshkumar C, Rajarajan G (2014) Optical and conductivity analysis of thiourea single crystals. Rasayan J Chem 7:287–294
Isakov H, Askarov IR, Usmanov S (2018) FTIR spectroscopic studies of compounds of thiourea-formaldehyde oligomers. Universum: Eng Sci J 12(57) (in Rus)
Larkin P (2017) Infrared and Raman spectroscopy: principles and spectral interpretation. Elsevier
Ajayi TJ, Shapi M (2020) Solvent-free mechanochemical synthesis, hirshfeld surface analysis, crystal structure, spectroscopic characterization and NBO analysis of Bis (ammonium) Bis ((4-methoxyphenyl) phosphonodithioato)-nickel (II) dihydrate with DFT studies. J Mol Struct 1202:127254
Glendening ED, Reed AE, Carpenter JE, Weinhold F (1998) NBO Version 3.1, TCl. University of Wisconsin, Madison
Bader RFW (1990) Atoms in molecules – a quantum theory. Oxford University Press, Oxford
Rozas I, Alkorta I, Elguero J (2000) Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J Am Chem Soc 122(45):11154–11161
Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W (2010) Revealing noncovalent interactions. J Am Chem Soc 132(18):6498–6506
Agarwal P, Bee S, Gupta A, Tandon P, Rastogi VK, Mishra S, Rawat P (2014) Quantum chemical study on influence of intermolecular hydrogen bonding on the geometry, the atomic charges and the vibrational dynamics of 2,6-dichlorobenzonitrile. Spectrochim Acta A Mol Biomol Spectrosc 121:464–482
Niu X, Huang Z, Ma L, Shen T, Guo L (2013) Density functional theory, natural bond orbital and quantum theory of atoms in molecule analyses on the hydrogen bonding interactions in tryptophan-water complexes. J Chem Sci 125(4):949–958
Acknowledgments
The devices of the Krasnoyarsk Regional Center of Research Equipment of Federal Research Center “Krasnoyarsk Science Center SB RAS” were used in the work. The authors are grateful to G.N. Bondarenko for obtaining X-ray data and Korolkova I.V. for obtaining FTIR spectra.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Akman, F., Issaoui, N. & Kazachenko, A.S. Intermolecular hydrogen bond interactions in the thiourea/water complexes (Thio-(H2O)n) (n = 1, …, 5): X-ray, DFT, NBO, AIM, and RDG analyses. J Mol Model 26, 161 (2020). https://doi.org/10.1007/s00894-020-04423-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04423-3