
Abstract
Muscovite (Ms) and phlogopite (Phl) belong to the 2:1 dioctahedral and trioctahedral layer silicates, respectively, and are the end members of Ms-Phl series minerals. This series was studied in the 2M1 polytype and modeled by the substitution of three Mg2+ cations in the Phl octahedral sites by two Al3+ and one vacancy, increasing the substitution up to reach the Ms. The series was computationally examined at DFT level as a function of pressure to 9 GPa. Cell parameters as a function of pressure and composition, and bulk moduli as a function of the composition agrees with the existing experimental results. The mixing Gibbs free energy was calculated as a function of composition. From these data, approximated solvi were calculated at increasing pressure. A gap of solubility is found, decreasing the gap of solubility at high pressure.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Micas are one of the most common types of layered hydrous silicates, which occur in diverse crustal rocks (igneous, metamorphic, and sedimentary rocks). They are typically formed as alteration products and help in transporting significant amounts of water into the Earths’ interior at subduction zones. Much of the water carried to great depth during subduction of sediments is bound in hydrous minerals and released through dehydration reactions that could occur from 100-km depth [1, 2]. White micas and the serpentine mineral antigorite have the capacity to transport water beyond arc front depths [3, 4]. Micas could act as geo-thermo-barometers and help for deciphering the thermal regime, depths, and stresses that the rocks were subjected to.
Muscovite (Ms) and phlogopite (Phl) are white micas, belonging to the 2:1 dioctahedral and trioctahedral layer silicates, with ideal end members’ compositions of [KAl2(Si3Al)O10(OH)2] and [KMg3(Si3Al)O10(OH)2], respectively. Micas incorporate many ions in their structures and show an extensive chemical composition by a wide variation of temperature and pressure [5, 6]. Therefore, pure end members compositions are unusual to be found in nature. In fact, their natural series minerals cannot be represented by only two end members, but by simultaneous isomorphous substitutions of different cations in the structure. Ms and Phl can contain VIAl3+ and VIMg2+ in their octahedral (VI) sheets (the first superscript indicates the coordination of the cations in octahedral sheet into the crystal), respectively. There are many works studying the extent of the series [5,6,7,8,9]. The incorporation of Mg2+ in the structure of Ms can be substituted by two mechanisms: (1) phengitic substitution and (2) phlogopite substitution. The first one preserves the dioctahedral character of Ms, by changing the substitution of Al3+ in the tetrahedral sheet; meanwhile, in the last one, Mg2+ substitution in the octahedral sheet leads to a trioctahedral mica, without any changing in the composition of the tetrahedral sheet [6]. The dioctahedral content into the trioctahedral phase was usually considered to be in the limit of 10–15% [7, 8]. While Ms can have phengitic composition, KAl1.5Mg0.5(Si3.5 Al0.5)O10(OH)2, by the Tschermak substitution, Phl can have moderate to high amounts of VIAl3+ concentrations, with composition KMg2Al(Si2Al2)O10(OH)2 [9]. Ms-Phl series was examined to low pressure (up to 2 kbar) and high temperature [5, 6]. At these conditions, Mornier and Robert [5, 6] found that the members of the series are a mixture of Phengite and Phl substitutions and there is scarcity of pure binary Ms-Phl members. However, a decrease of temperature produced an important increase in the extent of the Ms-Phl mixed member.
Ms mainly occurs in acid plutonic igneous rocks, and in low- and high-pressure metamorphism, Ms is the principal bearer of K in metapelites at conditions where the K-feldspar is not stable. At temperature of 298 K, and approximately at 18 GPa, Ms loses its crystallinity at long range and amorphizes at 27 GPa. At 800 °C, Ms has a limited pressure stability, at ~ 4 GPa, it transforms to an assemblage of sandinite, corundum (Al2O3), water, and a hydrated mineral called K-cymrite [10]. A series of phase transformations is further undergone eventually forming hollandite [10, 11]. Sekine et al. [12] suggest that Ms is stable to pressures larger than 10 GPa, and from Domanik and Holloway [13, 14], phengitic Ms breaks down beyond 11 GPa and 900 °C.
In general, hydrous minerals can even endure to depths > 300 km. Ms is one of the most stable minerals in subducted oceanic sediments [1]; in such a way, in environment without quartz and pore fluids, Ms could reach 130-km depth [1].
Based on experimental phase relations, Phl could be stable to 9 GPa [15, 16]. In the breakdown products of Phl, this mineral was still recovered at 9.0 GPa and 1365 °C [17] and has a pressure stability limit of 9–10 GPa at 1000 °C [18]. Natural Phl and Phl plus enstatite underwent high pressure and temperature and Phl was still recovered at 8 GPa and 1300 °C [19].
Phl can be found in eclogites in subduction slabs at an approximate depth of 200 km [20]. Approximately at this depth, dehydroxylation reaction could start [2]. However, the thermodynamic stability is affected when dioctahedral Ms substitutes trioctahedral Phl. This effect remains poorly understood and can provide us with valuable insight on how water could be efficiently transported into the Earth’s interior.
The determination of the behavior of the structure of micas at high-pressure of subductions zones, characterized by very low geothermal gradient parallel to the slab surface, is therefore critical in the understanding of the recycling of volatile, the genesis of arc magmas and the metasomatism of the upper mantle (e.g. [21]).
It is well known that micas have a significant anisotropy in their crystal structure, with strong bonding along the tetrahedral and octahedral layers, and relatively weaker bonding across the layers. It is anticipated that the weaker bonding across the layers will lead to significantly different compressibility in and across the layers [22]. How this effect varies across the Ms-Phl needs to be deeply explored.
Computational methods allow us to explore in detail the behavior of this complex series at high pressure. In spite of the limited solubility of the dioctahedral phase in the trioctahedral phase, it is interesting exploring how, at atomistic scale, the structure varies across a model of Ms-Phl series and influences the energetics and bulk compressibility. DFT methods have been extremely useful in elucidating the high-pressure behavior of Ms [23, 24], Phl [25], Muscovite-Paragonite (Ms-Pg) series [26], and the evolution of their elastic constants [27]. Besides, these methods have also proved to be very reliable for studying many different problems in phyllosilicates, some fruitful examples can be referenced, such as: the infrared spectra ([28], and references therein), cation arrangements [29], Brønsted sites on acid-treated montmorillonites [30], isomorphic substitution and adsorption of metals [31] and amino acids [32], the mechanism of dissolution [33] and dehydroxylation [34], order-disorder of cations ([35], and references therein), etc.
Taking into account, Ms and Phl are found in many different rocks, genetic processes, and depths, and taking into account that the majority of them are non-end members, but mixed members, it would be interesting to study the Ms-Phl series, with the aim to know how the pressure affects the mixture and understand how these minerals could be in rocks, in subduction slabs, either as end or mixed members at increasing depth in the Earth. Thus, a systematic computational DFT study of the influence of pressure on a model of Ms-Phl series is here undertaken, free of other cation substitutions different of Al3+ and Mg2+ in the octahedral sheet, in an attempt to study the crystal parameters, compressibility, stability and properties variation.
Methods
Models
The complex stoichiometry of the Ms-Phl series may be expressed by KAlx□x/2Mg3−3x/2 (Si3Al)O10(OH)2, where □ represent the octahedral vacancy, x = 0 represents for Phl end member and x = 2 represents for Ms end member. In the tetrahedral site the Si:Al ratio is always 3:1. This is primarily to attain the charge balance with the interlayer cation, a K+ in this case. K+ content and composition of tetrahedral sheet are here considered constant. Sometimes, it is necessary to distinguish between two types of octahedral sites, M1 and M2, with two M2 sites and one M1 site per half unit cell, and according to it, the Al3+ cations occupies the octahedral M2 sites, leaving the M1 sites mostly vacant, i.e., with an octahedral cluster configuration of □M1AlM2AlM2. The largest size of the M1 sites with respect to M2 sites [36], the smaller ionic radius of the Al3+ with respect to Mg2+, and the symmetry of the M2 sites, justify this choice. Besides, in the dioctahedral micas, the Al3+ is located in the M2 sites and the vacants are located in the M1 sites. In contrast, the Mg2+ cations occupy both the M1 and M2 sites of the octahedral sheet, with an octahedral cluster configuration of MgM1MgM2MgM2 (Figs. 1 and 2). Our models have been designed being always the three Mg2+ cations in the same octahedral cavity, for keeping the balance of charge in the octahedral sheet as localized as possible, and the two VIAl3+ cations and vacancy in the octahedral sheet substituting the cluster of three Mg2+ cations. The substitutions in different sheets are kept as far as possible. The disordering of cation configurations is not considered in this study due to the small size of the models. Hence, in this computational study, the energetic effect associated to different cationic configurations is neglected, which will be addressed in future works. All models of the Ms-Phl series were performed in the 2M1 polytype. The unit cells considered in this study have 84 atoms for the Ms end member [K4Al8□4(Si12Al4)O40(OH)8] and 88 atoms for Phl end member [K4Mg12(Si12Al4)O40(OH)8]. K is located in the interlayer space; next, Al3+, □, and Mg2+ are located in the octahedral sheet; Si4+ and Al3+ in brackets are in the tetrahedral sheet, O40 are oxygen in the octahedral and tetrahedral sheets; and OH are in the octahedral sheet in approximately the middle of the tetrahedral cavity (see Fig. 1 and 2). Five compositions are considered: Phl; X(Ms) = 0.25, 0.50, 0.75; and Ms (Fig. 2), where X(Ms) = ½x, x being the subindex in the general chemical formulae of the Ms-Phl series. The configurations at X(Ms) = 0.25 and 0.75 could be considered as interstratified configurations, since one octahedral sheet is a true dioctahedral/trioctahedral sheet, while the other sheet is either trioctahedral or dioctahedral, respectively.

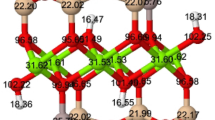
2M1 polytype for the Ms-Phl series for the member X(Ms) = 0.50. Blue, navy blue, and brown polyhedra represent the Al3+, Si4+, and Mg2+ cations, respectively. O and H atoms and K+ cation are represented by red, white, and purple balls, respectively

The different cationic configurations in two octahedral sheets of our models viewed at the (001) plane. Up and down figures correspond to up-octahedral sheet and down-octahedral sheet, respectively, of our model. The effective a-b cells are represented by fine blue lines in both octahedral sheets. From left to right: Ms, X(Ms) = 0.75, 0.50, 0.25, and Phl. Al3+ octahedra are represented by blue and navy blue octahedra, Mg2+ octahedra are represented in beige and brown octahedra, and O and H are represented by red and white balls
Theoretical methods
All calculations were performed at the DFT level. Two different codes were used: (1) SIESTA trunk-462 version [37,38,39] and (2) Quantum ESPRESSO 5.4 [40, 41].
Siesta calculations
Numerical atomic orbitals (NAO) at double ζ plus polarization basis sets, the generalized gradient approximation (GGA), and Perdew-Burke-Ernzerhof (PBE) [42] correlation-exchange functional were used. A mesh cut-off of 500 Ry and 15 k-points in the Monkhorst-Pack grid, along with Troullier-Martins (TM) norm-conserving pseudopotentials [43] were used. These calculations could be referred to as NAO+PBE+TM, but it can be shortened as NAO. The convergence thresholds of forces and stresses were 0.005 eV/Å and 0.001 GPa, respectively. To study the excess behavior, binding energies have been calculated. The basis sets and pseudopotentials were optimized for a set of dioctahedral 2:1 phyllosilicates [44, 45]. Ortega-Castro et al. [23] found that the pressure behavior of Ms calculated with the Local Density Approximation with the Ceperly-Alder correlation-exchange functional [46] yielded cell parameters and volumes in less agreement with the experimental results than the GGA results. GGA functionals also gave better results in kaolinites [47]. The dispersion forces corrections were analyzed in non-inter-layered charged phyllosilicates, finding its important role in these minerals [48]. However, in the Ms-Phl, the most important forces in the interlayer space are the Coulomb forces due to the type of substitutions in the tetrahedral sheet, which are well described by the DFT methods.
Quantum ESPRESSO calculations
The influence of basis set and dispersion forces was further studied by performing additional calculations with plane waves basis sets, PAW pseudopotentials [49], and the combination of the B86b [50] exchange functional with PBE [42] correlation functional, along with the exchange-hole dipole moment model (XDM) [51, 52]. The PAW data sets were generated from the pseudopotential library pslibrary [53]. The B86BPBE+XDM functional was proven to be a good choice to describe weak interactions [52]. This method is able to reproduce the crystal structure of layered hydrous silicates such as Ms and Phl and the intermediate compositions. These calculations will be referred to as PW+PB86BPBE+XDM+PAW but it can be abridged as PW. Some preliminary calculations were performed in order to obtain an optimal plane-wave kinetic energy cut-off, density cutoff, and k-point grid, being 110 Ry, 440 Ry, and 5 × 3 × 5, respectively. The convergence thresholds of forces and stresses were 0.001 Ry/Bohr and 0.05 GPa, respectively.
Equation of state
Taking into account the range of stability of both end members, especially that of Phl (8 GPa [19] and 9 GPa [15,16,17,18]), the calculations were carried out at pressure from − 1 to 9 GPa. The negative pressures indicates that the cell volume is larger than the zero pressure cell volume, and hence represents a condition where traction stresses were considered. At the NAO level, the P increments of 0.25 GPa were used between − 1 and 1 GPa, a pressure increment of 1 GPa was used between 1 and 3 GPa, and at pressure greater than 3 GPa and up to 9 GPa, increments of 3 GPa were used. At PW level, structures at pressures of − 1, − 0.9, − 0.6, and − 0.3 GPa were calculated, and also a range of pressures from 0 to 9 GPa, with an increment of 1 GPa was used. The pressure and volume results were fitted to a third order Birch-Murnagham (BM3) equation of state:
where fv is the Eulerian finite strain, K0 refers to the bulk modulus at a pressure of 0 GPa, and K′ \( \left(=\frac{dK}{dP}\right) \).
We have fitted V0, the unit cell volume at a pressure of 0 GPa, bulk modulus (K0) and its first derivative (K′) with respect to pressure for the five members of the series. The program EOSFIT 5.2 [54, 55] was used to fit the pressure and volume data.
Mixing Energetics
The mixing Gibbs free energy, ΔGmix, is given by the following:
where Gxs is the excess Gibbs energy, which gives the deviation of a solution from the mechanical mixture. Then Gxs was calculated at a fixed pressure and temperature with the formalism:
where the index i is for an intermediate compositions of the mixture, the index j represents a chosen end member of the series, and Xij is the molar fraction of the j end member in the mixed member; ΔGideal is the free energy for the entropy change of the random mixing of the cations. Gxs,i can be described following a Redlich-Kister equation [56,57,58]:
where X is the molar fraction of one component in the binary series, and each A, B, and C coefficients are dependent of temperature and pressure, and they can be resolved in enthalpy, entropy, and volume components as follows [59]:
similar equations are obtained for B and C coefficients of Eq. (4). AH can be calculated from the binding energy. Slaughter [60] and Yu [61, 62] found that the binding energy is especially suitable for calculating the excess enthalpy in silicates. This last author introduced a scale factor related with the experimental enthalpy of formation of the end members [63]. In our case, scaled factors (fj) with the experimental heat of formation of Ms and Phl [64] are calculated:
where j is an end member; and an interpolated scaled factor between the end member scaled factors are used for the mixed members.
From the total energy of the DFT methods, the binding energy can be calculated, and from this the excess energy can be also calculated with Eq. (6) and an equation similar to Eq. (3):
The Sxs is considered null in the same way that the model D of ref. [59] for the Ms-Pg series, and the corresponding coefficients in Eqs. (4) and (5) are null. So, the entropic effects are transferred to ΔGideal. The excess volume (Vxs,i) can be obtained from an equation similar to Eqs. (3) and (7).
ΔGideal is calculated with the following equation:
where aj is the activities of the end members, in the composition of the mixed members, which are given by Price [65] and Yu’s equations [61, 62] and the mixing on-site model [66], and adapted to our systems:
The ΔGideal is a measure of the change of configurational entropy of cations as a consequence of the random mixture in their corresponding crystallographic sites. Of course, the small size of our model makes it difficult to explore random cationic configurations, so Eqs. (9) and (10) could be considered as a useful approximation for knowing how the ideal entropy can affect the excess Gibbs free energy of our system, adding it to our excess binding energy and taking into account all reservations and cautions that this approach has.
Taking into account the approximations of the Gxs and Gideal, the mixing Gibbs free energy (Eq. 2) can be fitted by the least square methods to a quartic polynomium in X(Ms) at constant temperature and pressure:
From the first and the second derivative with respect to composition, we can calculate the positions of the minima and saddle points of Eq. (11). With the two minima and saddle points compositions at different temperatures and constant pressure, the binodes and the spinodes of a Gmix-solvus curve can be built.
Results and discussion
Crystal structure
At a pressure of 0 GPa, in general, the unit cell parameters of Phl and Ms are in agreement with the range of experimental data (Table 1; computational data are compared with different authors’ data [22, 23, 25,26,27, 67,68,69,70,71,72,73,74,75,76,77,78,79]), decreasing as a function of the Ms content in the series (Table 1 and Fig. 3). Our results are consistent with previous results of Phl at 1 M polytype from the plane-wave LDA and GGA calculations [25]. Cell parameters as a function of composition, X(Ms), are fitted to quadratic polynomials (Fig. 3).

Cell parameters variation (a, b, and ½csinβ) as a function of X(Ms). B86 means PW
Average Si-O and IVAl-O bond distances are in very good agreement with the experimental results, with a slight mismatch at the Ms end-member (Table 1). The octahedral bond distances are also in the range of the experimental values. They are different depending on the cation, and, in general, the VIMg2+/VIAl3+-O distances tend to decrease from Phl to Ms across the series. The experimental octahedral Al-OH bond length for Ms (1.914 Å [74]) agrees with the computational range (1.914–1.919 Å at NAO level). Other interesting geometrical features are the distances from the K+ in the interlayer space to the closest and farthest basal oxygen atoms, inner and outer distances referred to as d(K··O)in and d(K··O)out, respectively. Our calculated d(K··O)out values are slightly larger, and the d(K··O)in slightly shorter than the experimental values. The α angle is defined as the average rotation of the tetrahedral units along the apical axis, quantifying the deviation from the ideal hexagonal symmetry of the tetrahedral cavity. The α angle for the end members are larger compared to the range of experimental values reported, and increases from Phl to Ms. The volumes of IVSi/IVAl units decrease along the series (2.317–2.273 Å3/2.874–2.774 Å3) and agrees with the experimental value (2.334 Å3 [22]; [67,68,69]), and the VIMg/VIAl volume units decrease from Phl to Ms across the series (11.62–11.440 Å3 /9.555–9.3386 Å3), according to the cationic ionic radii. The tetrahedral sheet thickness shows similar values in Phl and Ms and is also similar to the experimental results. The octahedral sheet thickness is larger in Phl than in Ms, and the value of Ms is close to the experimental value. Finally, the interlayer thickness is the largest one in the crystal structure, and Phl shows a larger value than the Ms.
Crystal structure at increasing pressure
The effect of pressure on the unit cell parameters for Ms has been evaluated in the Ms-Pg series minerals [26] (see Electronic Supplementary Material). Their theoretical values are in agreement with the known experimental values.
The effect of pressure on the unit cell parameters for Phl is reported in Fig. 4. The length of the a axis decreases from 0 to 9 GPa. The effect of pressure on a axis can be described by a linear trend (Fig. 4a), and agrees very well with all known experimental values, especially with the Hazen and Finger’s results [22]. Other experimental results are also in good agreement, but with slightly different slopes, resulting in a crossover of the pressure dependence of the length of the a axis ~ 3 GP [67]. Some other experimental results have very similar slopes but slight different values [68]. The a values as a function of pressure calculated using PW match the NAO values at low pressure and are slightly smaller at high P. Therefore, we can expect the compressibility of the a axis at the PW level be larger than the NAO compressibility. The behavior of the b axis shows approximately the same trends (Fig. 4b).

a–d Cell parameters and volume of Phl as a function of pressure. Comparisons with the values of Hazen & Finger 1968 [22], Pavese et al. 2003 [67] and Comodi et al. and Comodi et al. 2004 [68] (two samples, for the second sample a “II” is added). c values are plotted as ½csinβ and for the other authors values are plotted as csinβ; our volume is plotted as ½ V, and for the other authors volume is plotted without any treatment
To compare with 1 M polytypes we used ½csinβ and our results are very close to the previous experimental results (Fig. 4c). Notice that our values of the β angle (94.5–94.9° and 94.6–94.7° from − 1 to 9 GPa at NAO and PW level, respectively (see Electronic Supplementary Material) are lower than the β angle reported in the 1M polytypes (100.0–100.74° [22]). Despite a maximum discrepancy of ~ 5° in the angular lattice parameter, our theoretical results show similar pressure dependence as the experiments. The variation of ½csinβ as a function of pressure (Fig. 4c) could be described with a quadratic function, and its magnitude decreases at NAO between 0 and 9 GPa. ½csinβ is one of the softest directions in the crystal structure, which is affected by the weakest forces of the crystal at the interlayer region. As expected, the crystal structure of micas is going to be most compressible along its weakest direction, which is perpendicular to tetrahedral-octahedral-tetrahedral layers, as it occurs in most of the layered materials. Our results are also in good agreement with the experimental values [67, 68]. The results from PW are approximately the same that the NAO results; however, at high pressure, a slightly larger compression is observed at PW with respect to NAO.
The effect of pressure on volume (see Electronic Supplementary Material) is considered with the half of the unit cell volumes (Fig. 4d) for comparing our results on 2M1 polytypes with the experimental results on 1M polytypes. The mineral volume reduces between 0 and 9 GPa. Results from PW show similar behavior to NAO calculations, being very close to the traction values. Notice that at high pressure the PW volumes show larger compression than the calculated with NAO. The PW results are quite close to the experimental results [67].
Hence, it is evident that, the computational methods employed in this study capture the existing experimental results on the crystal structure and the compression behavior of the end members Ms and Phl.
Bulk and linear moduli across the Ms-Phl series minerals
The variation of the bulk modulus, K0, and its derivative with respect to pressure, K′, as a function of X(Ms) show a linear behavior in NAO calculations (Table 2, and Fig. 5; data are compared with different experimental data [10, 12, 22,23,24, 56, 64, 67, 68, 74, 79,80,81]). K0 increases (Fig. 5a), whereas K′ decreases (Fig. 5b) as a function of X(Ms). The K0 for Ms is greater than Phl by 6 GPa. The K0 values calculated at PW are lower than NAO values by 6 GPa. The behavior of K0 vs X(Ms) from PW shows a quadratic behavior (Fig. 5a). The experimental values of the end members show a greater dispersion. Nonetheless, the NAO values of K0 are close to the experimental values, which are on the higher end of the experimental range, and the PW values are close to those which are at the lower end of the experimental scatter.

a K0, b K′ and K″, and c K0csinβ, K′0csinβ, and K″0csinβ as a function of X(Ms). In the inset, K means K0 at NAO and PW; Pave03, Pavese et al. (2003) [67]; Como04, Comodi et al. [68]; H&F78, Hazen and Finger [22]; C&Z95, Comodi and Zanazzi [79]; V&G86, Vaughan and Gugenheim [81]; F&N94, Faust and Knittle [10]; Cat94, Catti et al. [74]; U&V15, Ulian and Valdrès [24]; either H&P11 or HP11, Holland and Powell (2011) [64]. The K′ and K″ before the previous acronyms mean that they are extracted from the corresponding references
The K′ experimental values of Ms also show a great scatter from 3.2 to 6.9, but those of Phl are less spread out (Table 2, Fig. 5b). Nonetheless, K′ values are also around the experimental results, decreasing from the Phl end member to Ms end member. K′ of this series is high by comparison with other more packed minerals [82]. K′ of the Phl members are larger than the Ms members (ΔK′Phl-Ms~1.9). Thus, the bulk modulus of Phl end and mixed members increases at a greater rate with compression than the Ms mixed members (or, in other words, the amount of VIMg2+ does that K0 increases at greater rate with pressure). Both methods yield the same trends, although K′ at NAO level is fitted to a linear function, and the PW results cannot fit to any polynomial function.
K″ values are negative, increasing with X(Ms), being close to the known experimental values (Table 2, Fig. 5b). The calculated K″ values also yields the same trend by both methods and agree with the experimental trend.
Cell parameter moduli at NAO level (Table 2), K0i, show the largest values for the a and b axis. Computational results are around twice larger than the experimental results. They do not show a clear functional form along the series. K0csinβ shows the smallest values of K0i and is around seven times smaller than K0a for Phl and Ms, indicating a great anisotropy (Ms, [79]; Phl, [22]). K0csinβ is very similar to the experimental values. K0csinβ, K′0csinβ and K″0csinβ in the series follows similar values and trends (Fig. 5b) than the bulk modulus values, indicating that the csinβ direction determines the stiffness of the whole structure.
Therefore, the [001] direction is the weaker direction in the structure and is responsible of its marked anisotropy [22, 67, 68]. This is due to the large value of the ionic radius of the K+ cation, its structural environment in the crystal structure, and the occurrence of the weakest forces at the interlayer space in the structure. As seen, the substitution of Mg2+ by Al3+ in the octahedral sheet also contributes to the anisotropy.
Approached solvus behavior of the series minerals
ΔGmix at 298 K and different pressures are calculated (Fig. 6a, NAO). These curves show two minima and a maximum at the central composition. The minima are situated near of the end member compositions and at equidistant molar fractions of the end members, Ms0.08Phl0.92 and Ms0.92Phl0.08, and the maximum at Ms0.5Phl0.5. The heights of the maxima decrease with the increasing pressure. When the temperature increases (673 K, Fig. 6b), the minima are deeper and the maxima lower down with respect to the previous ΔGmix298K curves, but the trend with respect to the pressure is maintained. Finally, at a higher temperature 1073 K (Fig. 6c), the behavior is the same, though at high pressure (9 GPa), the central maximum disappeared and a flat minimum is found. From these data an approach solvus from ΔGmix can be extracted (Fig. 7). At 0 GPa and 298 K, the solvus shows a wide gap of solubility, which is gradually closing to the consolute point at high temperature (Fig. 7). Rutherford [7] showed that the mineral annite (resulting from VIFe2+ substitution by VIMg2+ in Phl) cannot contain more than 10–15% of Ms. Ferraris et al. [83] found crystals of Phl exsolving from a crystal matrix of Ms in pegmatites, indicating the especial stability of the end members of the series at low pressure and the gap of solubility found in rocks of low pressure. The binodes and spinodes narrow the gap of solubility at high temperature and pressure, keeping the compositions at low temperature unchanged. The binodes show a greater change with pressure than the spinodes, in such a way that at 1073 K and 0 GPa, the difference of X(Ms) between the binodes is 0.56 and the spinodes is 0.32; however, at the same temperature and 3 GPa, the difference between binodes is 0.50 and between the spinodes is 0.30, which indicates a larger variation of ΔGmix with the pressure at the minima positions than at the saddle points. Besides, it is possible to extract from Fig. 7 that the position of the consolute temperature is also decreasing with pressure. All of these indicate a narrower gap of solubility at high pressure, but at low pressure and temperature the gap of solubility is the same. Robert [5] did not find any complete solution between dioctahedral and trioctahedral Phl micas at 600 °C and 0.1 GPa. Nonetheless, the micas found by this author had a greater IVAl3+ substitution that our theoretical models. At T > 873 K the trioctahedral substitution persists instead of the phengitic substitution, and the micas with our tetrahedral composition seem to be the most stable [6]. All of these could indicate the importance of different cations other than Al3+ and Mg2+ in the octahedral sheet. The Ms end member breaks down at approximately 4 GPa and 1073 K [10]. So, at first sight, it could be thought the Ms end member is not relevant from 1000 K and moderate pressure in the subduction slabs. On the other hand, Phl end member is stable without breaking down at higher pressure and temperature (9 GPa and 1638 K [17], 9–10 GPa and 1273 K [18], 8 GPa and 1673 K [19]) than Ms. With the increasing solubility of Ms at high pressure and temperature, and with available Mg2+, the system would go to a mixed member with more Phl in the structure, which would allow the system to be more resistant to temperature and pressure than the Ms end member and reaches deeper depths in the upper mantle of the Earth. Nonetheless, our calculations have been performed with many approximations: (1) the different configurations in the natural rocks, which have not been taken into account in our calculations; (2) all the temperature effects in these solvi are approached from the ideal configurational Gibbs free energy; and (3) Gxs have been obtained at 0 K, without taking into account any vibrational effects, with a small crystal model and considering only fixed cationic configurations. Nevertheless, the theoretical Gxs comes from a model where there is an energetic difference between the binding energy of the mixed member composition minus a model of the mechanical mixture with the same composition than the mixed member. In this difference, many vibrational effects could be canceled.

Mixing Gibbs free energy (kJ/mol) at NAO as a function of X(Ms) at different pressures: a 298 K, b 673 K, and c 1073 K. Energies in kJ/mol per formulae of KAlx□½xMg3-(x+½x)(Si3Al)O10(OH)2

Solvi at NAO at different pressures. S1 and S2 binodes at low and high X(Ms), respectively. SP1 and SP2 spinodes at low and high X(Ms), respectively
At PW, ΔGmix shows a qualitatively similar behavior (Fig. 8a–c) to NAO, and the pressure effect is less noticeable than NAO, corresponding to lower differences of GxsPW than GxsNAO. At 1673 K, the maximum is still maintained at 9 GPa (Fig. 8c). Besides, the minima show different depths, the minimum at the side of the Phl end member being deeper than at the side of the Ms end member. The solvi (Fig. 9) indicate consolute temperatures higher than the previous NAO consolute temperatures, starting to narrow the gap of solubility at higher temperature and being asymmetric than those from NAO. The change of two Al3+ plus an empty cavity by three Mg2+ along with the greater size of the M1 sites with respect to M2 sites [36] could justify the asymmetry between minima and to be obtained at PW. In this case, the binodes at high P vary more than the spinodal curves at high pressure and temperature. Anyway, the behavior could be considered qualitatively similar to that of NAO, but the pressure effect is less perceptible and the temperature is noticeably higher. So, taking into account these last calculations, the pressure effect could be led to be relevant at higher depths and temperature inside the Earth’s mantle.

Mixing Gibbs free energy (kJ/mol) at PW as a function of X(Ms) at different pressures: a 298 K, b 1073 K, and c 1673 K. Energies in kJ/mol per formulae of KAlx□½xMg3-(x+½x)(Si3Al)O10(OH)2

Solvi at PW at different pressures. S1 and S2 binodes at low and high X(Ms), respectively. SP1 and SP2 spinodes at low and high X(Ms), respectively
Our results indicate that these materials should have compositions near the end members of the Ms-Phl joint at low pressure. At increasing pressure and temperature, the mixing compositions are being much more probable.
In subduction slabs, it is possible to find Ms-bearing schists, which can subduct to 130-km depth, corresponding to a pressure of approximately 4 GPa. By sinking deeper, Ms could increase mixed compositions in the Ms-Phl series, so the extension of the mixed members would be larger. The increasing VIMg2+ content by the increasing depth would make the mineral more stable. Phl may occur in eclogites in subduction zones at larger depths, and the increasing pressure could produce mixed members with increasing dioctahedral composition if Al3+ would be available. If the rocks go to increasing temperature zones, the temperature coming from the shear heat or isotherms of high temperature inside the Earth’s mantle, the extension of the mixed compositions would be also favored.
When the magma rises from deep to shallow zones, the recrystallization of the different minerals are subjected to the available elements in the melt and the changing pressure and temperature, but if Al3+ is lacked in the melt, Phl end members would be the most probable.
The high temperature will favor the dehydroxylation reaction in phyllosilicates. This reaction comes from two VIOH reacting to yield one H2O molecule. The local cation environment around VIOH reactants changes the free energy barriers of the dehydroxylation reaction. In previous works, we studied by quantum mechanical methods the dehydroxylation reaction in pyrophyllite and smectite models [84,85,86,87,88,89]. We computationally found two reaction mechanisms for reacting two OH’s in the octahedral sheet: cross and on-site mechanisms. In the former, the proton of one OH goes through the octahedral cavity to react with other OH in the same octahedral cavity, and in the second mechanism the reaction is produced between two contiguous octahedra. In the two last works [88, 89], we also found that the increasing content of Mg2+ in dioctahedral smectites facilitates the dehydroxylation reaction. In the case of our Ms-Phl series in the subductions slabs, the increasing content of Mg2+ in Ms should also impact on the dehydroxylation reaction, possibly in the same direction than dioctahedral smectites. The increasing pressure producing mixed compositions and increasing the trioctahedral character of the octahedral layer, the cross mechanism will be decreased. Therefore, in the intermediate compositions of the Ms-Phl series, the possibility of a dehydroxylation reaction would decrease with respect to the Ms end member by inhibiting the cross mechanism, and increase with respect to the Phl end member by the opposite effect. Therefore, the mixed members will have different dehydroxylation conditions than their corresponding end members.
Conclusions
The calculated crystal structure, unit cell axes, and bulk moduli agree with the known values of the series. All of them have either linear or quadratic behavior as a function of X(Ms). Both NAO and PW results yield very similar cell parameters. At increasing pressure, the compression of the crystal structure is mainly accommodated by the shortening of the interlayer space. Consequently, the reduction in the c axis is significantly greater than the a and b axes. A great anisotropy is observed across the series, being more marked for the Phl.
The K0 of Phl end members of series are smaller than that of Ms end members. PW and NAO results are both close to the known experimental results, with both results having the same trend in the series. K′ and K″ results are also close to the known experimental results. Kcsinβ modulus increases and determines the stiffness of the bulk along the series.
The mixing Gibbs free energy calculated by NAO and PW shows quantitatively different values; however, the qualitative behavior for the series is the same. A gap of solubility is found without change at low temperature being independent of pressure; however, a decreasing of the gap of solubility and the critical temperature at high pressure is observed. Thus, we can expect the crystalline solubility of the central members of the Ms-Phl series to be larger at high pressure and temperature, which would yield important implications in the properties of these materials.
References
Huang WL, Wyllie PJ (1973) Muscovite dehydratation and melting in deep crust and subducted oceanic sediments. Earth Planet Sci Lett 18:133–136
Hwang H, Seoung D, Lee Y, Liu Z, Liermann HL, Cynn H, Vogt T, Kao C-C, Mao H-K (2017) A role for subducted super-hydrated kaolinite in Earth’s deep water cycle. Nat Geosci 10:947–953
Schmidt M, Poli S (1998) Experimentally based water budgets for dehydrating slabs and consequences for arc magma generation. Earth Planet Sci Lett 163:361–379
Schmidt M, Wielzeuf D, Auzanneau E (2004) Melting and dissolution of subducting crust at high pressures: the key role of white mica. Earth Planet Sci Lett 228:65–84
Robert J-L (1976) Phlogopite solid solutions in the system K2O-MgO-Al2O3-SiO2-H2O. Chem Geol 17:195–212
Monier G, Robert J-L (1986) Muscovite solid solutions in the system K2O-MgO-FeO-Al2O3-SiO2-H2O: an experimental study at 2 kbar PH2O and comparison with natural li-free white micas. Mineral Mag 50:257–266
Rutherford MJ (1973) The phase relations of aluminous iron biotites in the system KAlSi3O8-KAlSiO4-Al2O3-Fe-O-H. J Petrol 14:159–180
Foster MD (1960) Interpretation of the composition of trioctahedral micas. US Geol Surv Prof Pap 354:8, 48 pp
Crowley MS, Roy R (1964) Crystalline solubility in muscovite and phlogopite groups. Am Mineral 49:348
Faust J, Knittle E (1994) The equation of state, amorphization and high pressure phase diagram of muscovite. J Geophys Res 99:19785–19792
Mookherjee M, Steinle-Neumann G (2009) Deeply subducted crust from the elasticity of hollandite. Earth Planet Sci Lett 288:349–358
Sekine T, Rubin AM, Ahrens TJ (1991) Shock wave equation of state of muscovite. J Geophys Res 96:19675–19680
Domanik KJ, Holloway JR (1996) The stability and composition of phengitic muscovite and associated phases from 5.5 to 11 GPa. Implications for deeply subducted sediments. Geochim Cosmochim Acta 60:4133–4150
Domanik KJ, Holloway JR (2000) Experimental synthesis and phase relations of phengitic muscovite from 6.5 to 11 GPa in a calcareous methapelite from the Dabie mountains, China. Lithos 52:51–77
Yoder HS, Eugster HP (1954) Phlogopite synthesis and stability range. Geochim Cosmochim Acta 6:167–185
Yoder HS, Kushitro I (1969) Melting of a hydrous phase: phlogopite. Am J Sci 267(A):558–583
Kushiro I, Akimoto S, Syono Y (1967) Stability of phlogopite at high pressure and possible presence of phlogopite in the earth’s upper mantle. Earth Planet Sci Lett 3:197–203
Trønnes RG (2002) Stability range and decomposition of potassic richterite and phlogopite end member at 5-15 GPa. Mineral Petrol 74:129–148
Sato K, Katsura T, Ito E (1997) Phase relations of natural phlogopite with and without enstatite up to 8 GPa: implications for the mantle metasomatism. Earth Planet Sci Lett 146:511–526
Wyllie PJ, Sekine T (1982) Formation of mantle phlogopite in subduction zone hybridization. Contrib Mineral Petrol 79:375–380
Gill J (1981) Orogenic andesites and plate tectonics. Springer-Verlag, p 390
Hazen RM, Finger LW (1978) The crystal structures and compressibilities of layer minerals at high pressure. II Phlogopite and Chlorite. Am Mineral 63:293–296
Ortega-Castro J, Hernández-Haro N, Timón V, Sainz-Diaz CI, Hernández-Laguna A (2010) High-pressure behaviour of 2M1 muscovite. Am Mineral 95:249–259. https://doi.org/10.2138/am.2010.3035
Ullan G, Valdrè G (2015) Density functional investigation of the thermos-physical and thermo-chemical properties of 2M 1 muscovite. Am Mineral 100:935–944
Chheda TD, Mookherjee M, Mainprice D, dos Santos AM, Molaison JJ, Chantel J, Manthilake G, Bassett WA (2014) Structure and elasticity of phlogopite under compression: geophysical implications. Phys Earth Planet Inter 233:1–12
Hernández-Haro N, Muñoz-Santiburcio D, Pérez del Valle C, Ortega-Castro J, Sainz-Díaz CI, Garrido CJ, Hernández-Laguna A (2016) Computational study of pressure behaviour to 6 GPa of the 2M1 muscovite-paragonite series. Am Mineral 101:1207–1216
Hernández-Haro N, Ortega-Castro J, Pérez del Valle C, Muñoz-Santiburcio D, Sainz-Díaz CI, Hernández-Laguna A (2013) Computational study of the elastic behaviour of the 2M1 muscovite-paragonite series. Am Mineral 86:651–664
Hernández-Haro N, Ortega-Castro J, Pruneda M, Sainz-Díaz CI, Hernández-Laguna A (2014) Theoretical study on the influence of the Mg2+ and Al3+ octahedral cations on the vibrational spectra of hydroxyl groups of 2:1 dioctahedral phyllosilicate models. J Mol Model 20:2402, 10 pages
Escamilla-Roa E, Hernández-Laguna A, Sainz-Díaz CI (2013) Cation arrengement in the octahedral and tetrahedral sheets of cis vacant polymorph of dioctahedral 2:1 phyllosilicates by quantum mechanical calculations. Am Mineral 98:724–735
Briones-Jurado C, Agacino-Valdés E (2009) BrOnsted sites on acid-treated montmorillonites: a theoretical study with probe molecules. J Phys Chem A 113:8994–9001
Wang Q, Zhu C, Yun J, Yang G (2017) Isomorphic substitutions in clay materials and adsorption of metals onto external surfaces: a DFT investigation. J Phys Chem C 121:26722–26732
Escamilla-Roa E, Huertas FJ, Hernández-Laguna A, Sainz-Díaz CI (2017) A DFT study of the adsorption of glycine in the interlayer space of montmorillonite. Phys Chem Chem Phys 19:14961–14971
Wang Q, Zhu C, Yun J, Hu Q, Yang G (2018) Compositional transformations as well as thermodynamics and mechanism of dissolution for clay minerals. Chem Geol 494:109–116
Sainz-Díaz CI, Escamilla-Roa E, Hernández-Laguna A (2004) Pyrophyllite dehydroxylation process by first principle calculations. Am Mineral 69:1092–1100
Palin EJ, Dove MT, Redfern SAT, Ortega-Castro J, Sainz-Díaz CI, Hernández-Laguna A (2014) Computer simulations of cations order-disorder in 2:1 dioctahedral phyllosilicates using cation-exchange potentials and Monte Carlo methods. Int J Quantum Chem:114:1257–1286. https://doi.org/10.1002/qua.24703
Bailey SW (1984) Crystal chemistry of the true micas. Mineral Soc Am Rev Mineral 13:13–60
Sánchez-Portal D, Ordejón P, Artacho E, Soler JM (1997) Density-functional method for very large systems with LCAO basis sets. Int J Quantum Chem 65:453–461
Artacho E, Sánchez-Portal D, Ordejón P, García A, Soler JM (1999) Linear-scaling ab-initio calculations for large and complex systems. Phys Status Solidi B 215:809–817
Soler JM, Artacho E, Gale JD, García A, Junquera J, Ordejón P, Sánchez-Portal D (2002) The SIESTA method for ab-initio order-N materials simulation. J Phys Condens Matter 14:2745–2779
Giannozzi P, Baroni S, Bonini, N., Calandra, M., Car, R., Cavazzoni, C., Ceresoli, D., Chiarotti GL, Cococcioni M, Dabo I, Dal Corso A, de Gironcoli S, Fabris S, Fratesi G, Gebauer R, Gerstmann U, Gougoussis C, Kokalj A, Lazzeri M, Martin-Samos L, Marzari N, Mauri F, Mazzarello R, Paolini S, Pasquarello A, Paulatto L, Sbraccia C, Scandolo S, Sclauzero G, Seitsonen AP, Smogunov A, Umari P, and Wentzcovitch RM (2009) QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21:395502
Giannozzi P, Andreussi O, Brumme T, Bunau O, Nardelli MB, Calandra M, Car R, Cavazzoni C, Ceresoli D, Cococcioni M, Colonna N, Carnimeo I, Dal Corso A, de Gironcoli S, Delugas P, DiStasio Jr RA, Ferretti A, Floris A, Fratesi G, Fugallo G, Gebauer R, Gerstmann U, Giustino F, Gorni T, Jia J, Kawamura M, Ko H-Y, Kokalj A, Küçükbenli E, Lazzeri M, Marsili M, Marzari N, Mauri F, Nguyen NL, Nguyen H-V, Otero-de-la-Roza A, Paulatto L, Poncé S, Rocca D, Sabatini R, Santra B, Schlipf M, Seitsonen AP, Smogunov A, Timrov I, Thonhauser T, Umari P, Vast N, Wu X, Baroni S (2017) Advanced capabilities for materials modelling with quantum ESPRESSO. J Phys Condens Matter 29, 465901
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868
Troullier N, Martins JL (1991) Efficient pseudopotentials for plane-wave calculations. Phys Rev B 43:1993–2006
Ortega-Castro J, Hernández-Haro N, Hernández-Laguna A, Sainz-Díaz CI (2008) DFT calculation of crystallographic properties of dioctahedral 2:1 phyllosilicates. Clay Miner 43:351–361
Ortega-Castro J, Hernández-Haro N, Muñoz-Santiburcio D, Hernández-Laguna A, Sainz-Díaz CI (2009) Crystal structure and hydroxyl group vibrational frequencies of phyllosilicates by DFT methods. J Mol Struct THEOCHEM 912:82–87. https://doi.org/10.1016/j.theochem.2009.02.013
Ceperley DM, Alder BJ (1980) Ground state of the electron gas by a stochastic method. Phys Rev Lett 45:566–569
White CE, Provis JL, Riley DP, Kearley GJ, van Deventer JSJ (2009) What is the structure of kaolinite? Reconciling theory and experiment. J Phys Chem B 113:6756–6765
Tunega D, Bučo T, Zaoui A (2012) Assessment of ten DFT methods in predicting structures of sheet silicates: importance of dispersion corrections. J Chem Phys 137:114105
Blöchl PE (1994) Projector augmented-wave method. Phys Rev B 50:17953–17979
Becke AD (1986) On the large-gradient behavior of the density functional exchange energy. J Chem Phys 85:7184
Becke AD, Johnson ER (2007) Exchange-hole dipole moment and the dispersion interaction revisited. J Chem Phys 127:154108
de-la Roza AO, Johnson ER (2012) Van der Waals interactions in solids using the exchange-hole dipole moment model. J Chem Phys 136:174109
Dal Corso A., Pseudopotential Periodic Table: From H to Pu (2014) Comput Mater Sci 95, 337
Angel RJ (2000) Equations of state. In: Hazen RM, Downs RT (eds) High-pressure and high-temperature crystal chemistry Review in Mineralogy and Geochemistry, vol 41, pp 35–60
Angel RJ, Gonzalez-Platas J, Alvaro M (2014) Eosfit7c and a Fortran module (library) for equation of state calculations (2014) Z. Kristallogr. 229(5): 405–419. http://www.ccp14.ac.uk/ccp/web-mirrors/ross-angel/rja/soft/
Guggenheim EA (1937) Theoretical basis of Raoult’s law. Trans Faraday Soc 33:151–159
Redlich O, Kister T (1948) Algebraic representation of thermodynamic properties and the classification of solutions. Ind Eng Chem 40:345–348
Ganguly J (2008) Thermodynamics in earth and planetary sciences. Springer, Heidelberg
Roux J, Hovis GL (1996) Thermodynamic mixing model for muscovite-paragonite solutions based on solutions calorimetric and phase equilibrium data. J Petrol 57:1241–1254
Slaughter M (1966) Chemical binding in silicate minerals. Geochim Cosmochim Acta 30:299–339
Yu J-Y (1994) Theoretical calculation of Gibbs free energy of mixing between phlogopite and eastonite. J Geol Soc Korea 30:578–590
Yu J-Y (1997) Theoretical calculation of Gibbs free energy of mixing biotite: phlogopite-annite-eastonite-siderophyllite system. Geosci J 1:179–188
Helgeson HC, Delany JM, Nesbitt HW, Bird DK (1978) Summary and critique thermodynamic properties of rock forming minerals. Am J Sci 278A:1–229
Holland TJB, Powell R (2011) An improved and extended internally consistent thermodynamic dataset for phases of petrological interest, involving a new equation of state for solids. J Metamorph Geol 29:333–383
Price JG (1985) Ideal site mixing in solid solutions, with an application to two-feldspar geothermometry. Am Mineral 70:696–701
Nordstrom DK, Munoz JL (1985) Geochemical thermodinamycs. Benjamin /Cummings Publishing Co. Inc., Menlo Park 0-8053-6816-7
Pavese A, Levy D, Curetti N, Diella V, Fumagalli P, Sani A (2003) Equation of state and compressibility of phlogopite by in-situ high-pressure X-ray powder diffraction. Eur J Mineral 15:455–463
Comodi P, Fumagalli P, Montagnoli M, Zanazzi PF (2004) A single-crystal study on the pressure behavior of phlogopite and petrological implications. Am Mineral 89:647–653
Scordari F, Schingaro E, Mesto E, Lacalamita M (2012) 2M 1 –phlogopite from Black Hills (South Australia): the first case of configurational polytype in micas. Am Mineral 97:2016–2023
Burnham CW, Radoslovich EW (1964) Crystal structure of coexisting muscovite and paragonite. Carnegie Inst Wash Year Books 63:232–236
Rothbauer R (1971) Untersuchung eines 2M1-muskovits mit neutronenstrahlen. Neues Jb Mineral Monat 1971:143–154
Guggenheim S, Chang Y-H, Koster van Groos AF (1987) Muscovite dehydroxylation: high-temperature studies. Am Mineral 72:537–550
Catti M, Ferraris G, Ivaldi G (1989) Thermal strain analysis in the crystal structure of muscovite 2M1 at 700 °C. Eur J Mineral 1:625–632
Catti M, Ferraris G, Hull S, Pavese A (1994) Powder neutron diffraction study of 2M1 muscovite at room pressure and at 2 GPa. Eur J Mineral 6:171–178
Guidotti CV, Mazzoli C, Sassi FP, Blencoe JG (1992) Compositional controls on the cell dimensions of 2M1 muscovite and paragonite. Eur J Mineral 4:283–297
Brigatti MF, Frigieri P, Poppi L (1998) Crystal chemistry of Mg-, Fe-bearing muscovites-2M 1. Am Mineral 83:775–785
Mookherjee M, Redfern SAT, Zhang M (2001) Thermal response of structure and hydroxyl ion of phengite-2M 1: an in situ, neutron diffraction and FTIRstudy. Eur J Mineral 13:545–555
Mookherjee M, Redfern SAT (2002) A high-temperature Fourier transform infrared study of the interlayer and Si-O-stretching region in phengite-2M 1. Clay Miner 37:323–336
Comodi P, Zanazzi PF (1995) High-pressure structural study of muscovite. Phys Chem Miner 22:170–177
Comodi P, Zanazzi PF (1997) Pressure dependence of structural parameters of paragonite. Phys Chem Miner 24:274–280
Vaughan MT, Guggenheim S (1986) Elasticity of muscovite and its relationship to crystal structure. J Geophys Res 91:4657–4664
Anderson OL (1995) Equation of state of solids for geophysical and ceramic science. Oxford University Press, Oxford monographs on geology and geophysics 0-19-505606-X
Ferraris C, Grobety B, Weissicken R (2001) Phlogopite exsolutions within muscovite: a first evidence for a higher-temperature re-equilibration, studied by HRTEM and AEM techniques. Eur J Mineral 13:15–26
Molina-Montes E, Donadio D, Hernández-Laguna A, Sainz-Díaz CI, Parrinello M (2008a) DFT research on the dehydroxylation reaction of Pyrophyllite. 1. First principle molecular dynamics simulations. J Phys Chem B 112:7051–7060
Molina-Montes E, Donadio D, Hernández-Laguna A, Sainz-Díaz I (2008b) DFT research on the dehydroxylation reaction of Pyrophyllite. 2. Characterization of reactants, intermediates and transition states along the reaction path. J Chem Phys A 112:6373–7383
Molina-Montes E, Timón V, Hernández-Laguna A, Sainz-Díaz CI (2008c) Dehydroxylation mechanisms in Al3+/Fe3+ dioctahedral phyllosilicates by quantum mechanical methods with cluster models. Geochim Cosmochim Acta 72:3929–3938
Molina-Montes E, Donadio D, Hernández-Laguna A, Parrinello M, Sainz-Díaz CI (2013) Water release from pyrophyllite during the dehydroxylation process explored by quantum mechanical simulations. J Phys Chem C 117:7526–7532
Muñoz-Santiburcio D, Kosa M, Hernández-Laguna A, Sainz-Díaz CI, Parrinello M (2012) Ab initio molecular dynamics study of the Dehydroxylation Reaction in a Smectite model. J Phys Chem C 116:12203–12211
Muñoz-Santiburcio D, Hernández-Laguna A, Sainz-Díaz CI (2016) Simulating the Dehydroxylation reaction in Smectite models by Car−Parrinello-like−born−Oppenheimer molecular dynamics and Metadynamics. J Phys Chem C 120:28186–28192
Acknowledgements
The authors thank the “Centro de Supercomputación de Galicia” (CESGA) and “Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC), Universidad de Granada” for providing the computing time. The authors are thankful to F. Muñoz-Izquierdo, J. Rodríguez-Fernández, and M. Mookherjee for fruitful suggestions. This work was supported by Spanish MCINN and European FEDER grants CGL2008-02850/BTE, FIS2013-48444-C2-2P, FIS2016-77692-C2-2P, and PCIN-2017-098 and by the regional agency “Junta de Andalucía” for the RNM-264, -363, and -1897 PAI-grants.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper belongs to Topical Collection QUITEL 2018 (44th Congress of Theoretical Chemists of Latin Expression)
Electronic supplementary material
ESM 1
(DOCX 32 kb)
Rights and permissions
About this article

Cite this article
Hernández-Laguna, A., Pérez del Valle, C., Hernández-Haro, N. et al. Compressibility of 2M1 muscovite-phlogopite series minerals. J Mol Model 25, 341 (2019). https://doi.org/10.1007/s00894-019-4218-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4218-x