
Abstract
The Zika virus has recently become a subject of acute interest after the discovery of the link between viral infection and microcephaly in infants. Though a number of treatments are under active investigation, there are currently no approved treatments for the disease. To address this critical need, we screened more than 7 million compounds targeting the NS2B-NS3 protease in an attempt to identify promising inhibitor candidates. Starting with commercially and freely available compounds, we identified six hits utilizing an exhaustive consensus screening protocol, followed by molecular dynamics simulation and binding energy estimation using MM/GBSA and MM/PBSA methods. These compounds feature a variety of cores and functionalities, and all are predicted to have good pharmacokinetic profiles, making them promising candidates for screening assays.

Virtual screen of potential Zika virus NS2B-NS3 protease inhibitors
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Zika virus is a mosquito-borne flavivirus that has been known since 1947, but historically only caused mild symptoms such as fever in about 20% of infected individuals and rarely caused serious complications [1]. Public concern changed drastically during the 2015 epidemic in South America, where a number of reports of neurological disorders and microcephaly were reported [2]. Zika infection has been linked to a number of effects, including congenital microcephaly, heart defects and Guillain-Barre syndrome [3,4,5,6,7]. Since the spate of recent outbreaks, a number of vaccines are advancing through the clinical pipeline [8]. However, there are expected to be significant challenges associated with a Zika vaccine, as flaviviruses have a history of promoting antibody-mediated enhancement of infection [9]. Until the development of a vaccine, small molecule therapeutics are a promising candidate as both a treatment and as a prophylactic.
The Zika virus contains a positive-sense single-stranded RNA genome that encodes a single 3419-amino-acid-long polypeptide, which is cleaved to form three structural proteins (capsid, pre-membrane and envelope) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) [10]. NS3 contains the catalytic triad, and along with the transmembrane NS2B (which anchors NS3 to the endoplasmic reticulum) together form the NS2B-NS3 protease that is critical to viral replication and survival [11]. Given the critical role played by NS2B-NS3, it is an attractive target for small molecule inhibitors. While a handful of inhibitors have been evaluated, none to date have had acceptable profiles for advancement through preclinical trials, underscoring the critical need for the discovery of new drug-like scaffolds [12,13,14,15,16]. In order to address this need, we decided to run a virtual screen of millions of commercially and freely available drug-like molecules in order to identify compounds that could be readily purchased and screened for inhibitory activity. By using commercially and freely available compounds, we hope to accelerate the screening of these compounds and make them widely available to teams that lack access to synthetic chemistry resources.
Methods
Protein crystal selection and preparation
Fortunately, a handful of crystal structures are currently available for the NS2B-NS3 protease. However, the crystallization of Zika NS2B-NS3 and other closely related homologs has been complicated by its heterodimeric structure and the fact that the NS2B polypeptide is flexible, often leading to a disordered open conformation. In contrast, structures with bound inhibitors show that NS2B wraps around the NS3 structure and makes contact with some inhibitors, forming a closed conformation [17,18,19,20,21,22]. Some solved structures have utilized an artificial linker to stabilize the complex, but this has been shown to introduce steric hindrance and alter inhibitor binding interactions [23]. Based on this information, we decided to utilize an unlinked structure of NS2B-NS3 in complex with a small molecule in the closed conformation (PDB ID: 5H4I). This structure is in the more relevant closed conformation and has already been shown to be a good model for binding small molecules, thus we felt this structure was best suited for docking and evaluating drug-like inhibitors (Fig. 1) [24]. A Ramachandran plot generated using the RAMPAGE assessment tool [25] also showed that no residues resided in the outlier region, underlining the high quality of the structure (see supplementary material).

Structure of the unlinked NS2B-NS3 protease (PDB ID: 5H4I). Teal NS3 chain, maroon NS2B chain
To prepare 5H4I for docking, the structure was downloaded as a PDB file from the Protein Data Bank (https://www.rcsb.org/structure/5h4I) and the waters, acetate ion, and crystalized inhibitor were removed. The docking grid was prepared using AutoDockTools (ADT) [26] using the center of the crystallized inhibitor as the center of the search space grid (x = −5.576, y = 8.208, z = −13.937), the spacing set to 1 Å, and the dimensions set to 22x16x22 in the x, y, and z dimension, respectively. These parameters were selected to ensure coverage of the entire binding surface within the grid.
Preparation of the ligand libraries
Our initial library was built utilizing the ZINC15 database, which contains more than 230 million compounds in 3D format and provides vendor information for all commercially available items [27]. This data set was refined by only selecting compounds that were commercially available and in stock; we then applied a series of Lipinski filters to further refine the virtual library (molecular weight = 250–500, log P < 5, number of rotatable bonds ≤7, polar surface area ≤ 150 Å2, H-bond donors ≤5, H-bond acceptors ≤10), removed known toxicophores and PAINS [28] scaffolds, adjusted the pH of all compounds to physiological conditions, then compiled the library in PDBQT format. The final compiled library (hereafter referred to as the ZINC library) contained a total of 7,038,391 compounds.
In addition to the ZINC library, we also compiled a second library of compounds that are freely available through the National Cancer Institute (NCI) Developmental Therapeutics Program’s Open Chemical Repository [29]. We combined the Plated 2007, Mechanism 3 and Diversity 5 sets into a single library of 112,102 compounds in PDBQT format (hereafter referred to as the NCI library). While this library is significantly smaller, all compounds are made freely available to the research community.
Screening protocol
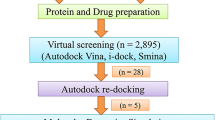
Our overall screening protocol for the ZINC library is outlined in Fig. 2.

Screening protocol of the ZINC library
After applying the Lipinski filters to the ZINC database, we screened the entire library utilizing both the Vina [26] and iDock [30] programs. The parameters for both programs were set such that each ligand had an average evaluation time of approximately 1 min based on initial timing tests. For the Vina program, the parameters were set to the following values: num_modes = 10, cpu = 6, exhaustiveness = 12. For the iDock program, the parameters were set to the following values: threads = 6, conformations = 10. All other program parameters were left at default values. A consensus scoring function was used to identify the top 1000 hits from this initial screen. These 1000 compounds were then re-screened again with both Vina and iDock using more exhaustive parameters designed such that each ligand had an average evaluation time of 1 h. For the Vina program, the parameters were set to the following values: num_modes = 20, cpu = 12, exhaustiveness = 1000. For the iDock program, the parameters were set to the following values: threads = 12, conformations = 20, trees = 5000, tasks = 640. The results from this exhaustive screen were re-scored again using the same consensus function. The top 25 hits were then visually inspected to ensure that the docking poses were reasonable and subsequently screened using a Tanimoto coefficient threshold of less than 0.4 based on the FP2 path-based fingerprint [31], resulting in the removal of nine compounds due to structural similarities. From the remaining 16 compounds, the top 10 were then examined using molecular dynamics (MD) simulations (described below) to confirm that the binding mode is preserved during equilibration and to estimate the binding energy, resulting in the final top 5 compounds with the most promising performance and scores. The top 1000 ranked consensus hits from our docking search against the ZINC database are provided in the supplementary material (Table S1).
The smaller NCI library was screened according to the protocol outlined in Fig. 3.

Screening protocol for the National Cancer Institute (NCI) library
The library was first screened with Vina and iDock using the same parameters and consensus scoring function as the initial ZINC library screen, but only the top 100 compounds were selected for the more exhaustive screen. After re-scoring, the top three compounds from the exhaustive screen were visually inspected and checked to ensure a Tanimoto coefficient of less than 0.4. These three compounds were then subjected to MD simulation followed by a binding energy determination, with the best performing compound selected as the single top hit for the NCI library. The top 100 ranked consensus hits from the NCI database are provided in the supplementary material (Table S2).
Consensus scoring
As is often the case when compounds are scored by different docking programs, we utilized a consensus scoring function to rank our results [32, 33]. For the initial screening of both libraries, the mean of the binding affinities were used to generate the top 1000 compounds from the ZINC library and the top 100 compounds from the NCI library. These subsets were then re-docked with the more exhaustive parameters described previously and the output binding affinities were then transformed by quantile normalization using MATLAB R2017a. The mean of the normalized energies for a compound then provided the basis for ranking. Previous work [20a] has suggested that this mean approach performs equivalently or better than other simple consensus methods using the median, maximum or minimum.
Molecular dynamics
MD simulations of the NS2B-NS3 complex with potential inhibitors identified by virtual screening were performed using Amber16 and AmberTools16 packages [34] with relative binding energies estimated from computed trajectories using molecular mechanics Poisson-Boltzmann surface area (MM/PBSA) and molecular mechanics generalized Born surface area (MM/GBSA) algorithms [35,36,37]. Topology files for the protein–ligand complex were constructed in TLEAP [34] using the FF14SB [38] and GAFF force fields [39]. The docked small molecule ligands were parameterized using ANTECHAMBER [34]. The complexes formed from the protein and the parameterized ligands were solvated in explicit TIP3P water [40], arranged in a truncated octahedron 8 Å from the surface of the protein. Prior to minimization, equilibration, and production run, the complexes were charge-neutralized by the addition of sodium ions.
The MD simulations were carried out using the particle mesh Ewald (PME) implementation of graphical processor unit–accelerated MD with the PME MD Compute Unified Device Architecture module of Amber16 [41]. Initially, the complexes were subjected to: (1) minimization with restraint of the protein for 1000 cycles; (2) unrestrained minimization for 1000 cycles; (3) equilibration while warming from 50 K to 300 K for 30 ps using a constant-volume periodic boundary condition; (4) equilibration with restraint of the protein at 310 K for 20 ps with a constant-pressure periodic boundary condition and using isotropic pressure scaling; and (5) unrestrained equilibration at 300 K for 500 ps. After minimization and equilibration, the complexes were subjected to a production run at 300 K for 50 ns. Preparation steps 1 and 2 comprised 500 steps of steepest descent, followed by 500 steps of conjugate-gradient descent. Steps 3–5 and the production runs used Langevin temperature regulation [42] with the collision frequency of 2.0 ps−1, bonds involving hydrogen were constrained by the SHAKE algorithm [43]; and a 12 Å cutoff was used for non-bonded interactions calculated by the PME method [44]. The MD simulations were monitored by examination of the internal energy and root mean standard deviation (RMSD) of the resulting trajectories (see Fig. S1 in the supplementary material). Production run trajectories were visualized using visual molecular dynamics (VMD; version 1.9.3) [45].
Binding energy computations
After completion of the production runs, binding energies were estimated using the MMPBSA.py module of AmberTools16 [35,36,37]. The simulations used the single trajectory method, and binding energies were calculated using the MM/PBSA and MM/GBSA algorithms [46]. The PBSA simulation used ionic strength of 0.15 mM (istrng = 0.150) and employed default radii from the prmtop file (radiop t = 0). The GBSA simulation used generalized Born “method two” (igb = 5) with 0.15 M salt concentration. We initially considered binding energy values over three 10-ps samples of a 50-ns MD-simulation, measured at 25 ns, 40 ns, and 50 ns (see Table S3 in the supplementary material). These observations suggested the estimated binding energies stabilized after 40 ns. As such, the binding energy of each protein–ligand complex was calculated utilizing the final 5 ns of each trajectory (Table 1).
Results and discussion
The molecular properties of the top five screening hits from the ZINC library (named ZC_01, ZC_02, etc.) and the top compound from the NCI library (NCI_01) are shown in Fig. 4, and selected molecular properties are listed in Table 1.

Structure of the top hits from the screening libraries
All selected compounds are calculated to have a high binding affinity, with binding energies ranging from −10.8 to −11.45 kcal mol−1. Each of the docking results was subjected to a MD simulation to examine the ability of the ligand to remain bound under equilibrating conditions, and also to provide an estimation of binding energy. Protein–ligand topologies were prepared and subjected to a 50 ns production run at 300 K. Qualitative examination of the protein–ligand structures during the trajectories show them to maintain stable complexes through various stages of the MD production run. For the complex of NS2-NS3B with ligand ZC_01, the RMSD increased slightly from 0 to 15 ns, presumably as the protein accommodated the ligand, and then relaxed into an apparently stable configuration from 15 ns to 50 ns with little change of RMSD in this latter part of the experiment (Fig. S1). For this representative example, the potential energy was also stable throughout the course of the 50 ns production run (Fig. S1). We found that the majority of the equilibration was complete within 10–20 ns for each of the protein–ligand complexes, making 50 ns production runs suitable for this system. Binding energies (ΔGbinding) were estimated using the MM/GBSA and MM/PBSA tools [46] and summarized in Table 1. While the MM/GBSA and MM/PBSA binding energies were measured by the respective algorithms, they were generally in agreement in terms of ranking. The predicted best compounds ZC-01, ZC-03, and ZC-05 had lowest predicted binding energies (−38.9 to −44.0 kcal mol−1 based on MM/GBSA; and − 28.2 to −35.7 kcal mol−1 based on MM/PBSA). The ΔGbinding values determined by the MM/GBSA and MM/PBSA methods provide a relative ranking that is fairly consistent with docking score, although the absolute binding energies should not be compared between these two algorithms. Across the series of compounds, the relative ranking produced by MM/GBSA appeared consistent compared with the MM/PBSA method. Potential inhibitor ZC_01 had the most favorable binding energy by the MM/GBSA and MM/PBSA methods. We observed that ZC_01, ZC_03, and ZC_05 had multiple H-bonding contacts with the putative binding site maintained through the course of the 50 ns MD simulation, and possibly contributed to their lower predicted binding energy. Qualitatively, these better scoring compounds also appeared to have complementary interactions with the hydrophobic protein residues at the binding site. Comparison of binding energies with a positive control inhibitor would be ideal, although currently a validated potent inhibitor at this binding site is not available. Given these limitations, we sought to identify inhibitors incorporated into novel scaffolds with low estimated binding energy in this study.
An additional ADMET analysis was performed on all compounds utilizing the admetSAR database [47] with the results listed in Table 2.
All compounds are predicted to have excellent absorption profiles and low oral toxicity, making them excellent starting candidates for small molecule therapeutic treatments. All compounds are also predicted to be non-carcinogenic, in addition to their low toxicity. It is also important to note that most compounds are predicted to have excellent permeability through the blood-brain barrier. It is known that the Zika virus can pass through the blood brain barrier, so any effective treatment would be expected to also share this property to effectively treat infected brain tissues [48].
Compound ZC_01 is a spiro compound and was calculated to have the highest binding affinity of all screened compounds that did not disassociate during MD simulations. Both docking programs predicted nearly identical binding poses, with a RMSD value of 0.197 Å (Fig. 5).

a Docking poses of ZC_01 calculated by Vina (peach backbone) and iDock (green backbone). RMSD = 0.197 Å. Grey NS3 peptide and interacting residues, magenta NS2B peptide and binding residues. b 2D molecular interaction map for ZC_01; dotted red lines polar interactions, green lines non-polar interactions
A number of hydrogen bonding interactions between the core of ZC_01 and nearby residues of NS3 are apparent, with two between the succinimide carbonyls with H51 and Y161, one between the backbone of G153 and the oxindole nitrogen, and one between the indole nitrogen and N152. One additional hydrogen bond between the pyrrolidine nitrogen and D33 of NS2B provides additional stabilization. Hydrophobic interactions are most apparent around the m-xylene moiety and are provided by A132 and Y161. Additional hydrophobic interactions are made by the nearby valine residues V72 and V155.
ZC_02 shares some similar interactions as with ZC_01, but is a much more synthetically attractive compound (Fig. 6).

a Docking poses of ZC_02 calculated by Vina (peach backbone) and iDock (green backbone). RMSD = 0.716 Å. Grey NS3 peptide and interacting residues, magenta NS2B peptide. b 2D molecular interaction map for ZC_02. Dotted red lines Polar interactions, green lines non-polar interactions
Again, the two docking poses predicted by Vina and iDock are very similar, with a RMSD value of 0.716 Å. The majority of hydrogen bonding interactions occur between the succinimide core and three NS3 residues, namely H51, S135 and Y161. An additional polar contact is made between Y150 and the m-trifluoromethylphenyl substituent, and hydrophobic interactions appear present from Y161, A143 and V72. Unlike ZC_01, however, ZC_02 does not appear to make any significant interaction with any NS2B residues.
Compound ZC_03 features a related 2,4-imidazolidinedione core in place of the succinimide core of ZC_01 and ZC_02, making contact with many of the same NS3 residues (Fig. 7).

a Docking poses of ZC_03 calculated by Vina (peach backbone) and iDock (green backbone). RMSD = 0.156 Å. Grey NS3 peptide and interacting residues, magenta NS2B peptide. b 2D molecular interaction map for ZC_03. Dotted red lines Polar interactions, green lines non-polar interactions
Specifically, hydrogen bonding interactions are apparent between the 2,4-imidazolidinedione moiety and H51 and N152. A naphthyl system resides in the hydrophobic pocket shared with V72, while the formamide substituted 1,4-benzoxazine makes polar contacts with S135, Y150 and Y161, with additional hydrophobic interactions provided Y161 and A132.
The somewhat lipophilic compound ZC_04 appears to bind primarily through hydrophobic interactions as expected, primarily with Y161, A132 and V72 (Fig. 8).

a Docking poses of ZC_04 calculated by Vina (peach backbone) and iDock (green backbone). RMSD = 0.122 Å. Grey NS3 peptide and interacting residues, magenta NS2B peptide. b 2D molecular interaction map for ZC_04. Dotted red lines Polar interactions, green lines non-polar interactions
ZC_04 also differs due to the fact that it has a triazolopyrimidine core, which appears to form a hydrogen bonding interaction with the S135 residue of NS3. Despite having fewer apparent polar interactions with the binding pocket than other inhibitors, both Vina and iDock predict a strong binding interaction (Table 1) and output nearly identical binding poses (RMSD = 0.122 Å).
Compound ZC_05 bears a urea core modified with an 1,4-diazepane and m-trifluoromethylphenyl system (Fig. 9).

a Docking poses of ZC_05 calculated by Vina (peach backbone) and iDock (green backbone). RMSD = 0.159 Å. Grey NS3 peptide and interacting residues, magenta NS2B peptide. b 2D molecular interaction map for ZC_05. Dotted red lines Polar interactions, green lines non-polar interactions
Similar to ZC_02, the trifluoromethylphenyl group makes polar contacts with the nearby S135 and Y150 residues, as well as hydrophobic interactions with A132 and Y161. The urea carbonyl also appears to make a polar contact with Y161. Additional hydrophobic interactions appear between residue V52 and the quinoxaline system.
The top compound from the NCI library, NCI_01, is a spiro molecule somewhat akin to the top hit for the ZINC library (ZC_01). A number of polar interactions are made between the barbituric acid ring and the nearby residues N152 and H51, as well as an interaction with the backbone of G153 (Fig. 10).

a Docking poses of NCI_01 calculated by Vina (peach backbone) and iDock (green backbone). RMSD = 0.064 Å. Grey NS3 peptide and interacting residue, magenta NS2B peptide. b 2D molecular interaction map for NCI_01. Dotted red linesPolar interactions are shown as dotted red lines, green lines non-polar interactions
The aromatic ring sits snugly in the hydrophobic pocket formed by Y161, A132 and Y150, with the Y161 residue also making an apparent hydrogen bonding interaction with cyclic ether oxygen atom. Both the Vina and iDock program predict nearly identical bonding poses for NCI_01, with a RMSD = 0.064 Å.
Conclusions
A collection of more than 7 million commercially and freely available compounds from the ZINC15 database and the NCI repository of small molecules were subjected to a virtual screening procedure consisting of consensus-based docking followed by MD simulation and binding energy calculations in order to identify promising potential inhibitors of the Zika NS2B-NS3 protease. The top five compounds from the ZINC library and the top compound from the NCI library are all predicted to be potent inhibitors of NS2B-NS3 and possess good pharmacokinetic profiles. These hits feature a number of different scaffolds and functional groups, representing a varied chemical space. To the best of our knowledge, this study is the largest in silico screen targeting a Zika protein. As efforts to develop treatment for Zika infection continue, there is a critical need to identify new, promising lead candidates. By selecting only compounds that are readily available, the hits disclosed in this study can be quickly screened without the need for synthetic preparation in an effort to rapidly identify new compounds for lead development.
References
Heymann DL, Hodgson A, Sall AA, Freedman DO, Staples JE, Althabe F, Baruah K, Vasconcelos PFC, Bino S, Menone KU (2016) Lancet 387:353–354. https://doi.org/10.1016/S0140-6736(16)00320-2
Russell PK (2016) PLoS Negl Trop Dis 10:e0004589. https://doi.org/10.1371/journal.pntd.0004589
Wilder-Smith A, Gubler DJ, Weaver SC, Monath TP, Heymann D, Scott TW (2017) Lancet Infect Dis 17:e101–e106. https://doi.org/10.1016/S1473-3099(16)30518-7
Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, Kolenc M, Us KR, Viptonik TV, Vodusek VF, Vizjak A, Pizem J, Petrovec M, Zupanc TA (2016) N Engl J Med 374:951–958. https://doi.org/10.1056/NEJMoa1600651
Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, Zhang N, Shi L, Qin C, Xu Z (2016) Cell Stem Cell 19:120–126. https://doi.org/10.1016/j.stem.2016.04.017
Cugola FR, Fernades IR, Russo FB, Freitas BC, Dias JLM, Guimaraes KP, Benazzato C, Almeida N, Pignatari GC, Romero S, Polonio CM, Cunha I, Freitas CL, Brandao WN, Rossato C, Andrade DG, Faria DP, Garcez AT, Buchpigel CA, Braconi CT, Mendes E, Sall AA, Zanotto PM, Peron JP, Muotri AR, Beltrao-Braga PCB (2016) Nature 534:267–271. https://doi.org/10.1038/nature18296
Vogel G (2016) Science 352:1375–1375. https://doi.org/10.1126/science.352.6292.1375
Fernandez E, Diamond MS (2017) Curr Opin Virol 23:59–67. https://doi.org/10.1016/j.coviro.2017.03.006
Malone RW, Homan J, Callahan MV, Glasspool-Malone J, Damodaran L, Schneider ADB, Zimler R, Talton J, Cobb RR, Ruzic I, Smith-Gagen J, Janies D, Wilson J (2016) PLoS Negl Trop Dis 10:e0004530. https://doi.org/10.1371/journal.pntd.0004530
Cox BD, Stanton RA, Schinazi RF (2015) Antivir Chem Chemother 24:118–126. https://doi.org/10.1177/2040206616653873
Kang C, Keller T, Luo D (2017) Trends Microbiol 25:797–808. https://doi.org/10.1016/j.tim.2017.07.001
Li Y, Zhang Z, Phoo WW, Loh YR, Li R, Yang HY, Jansson AE, Hill J, Keller TH, Nacro K, Luo D, Kang C (2018) Structure 26:555–564.e3. https://doi.org/10.1016/j.str.2018.02.005
Li Y, Zhang Z, Phoo WW, Loh YR, Wang W, Liu S, Chen MW, Hung AW, Keller TH, Luo D, Kang C (2017) Structure 25:1242–1250.e3. https://doi.org/10.1016/j.str.2017.06.006
Lei J, Hansen G, Nitsche C, Lkein CD, Zhang L, Hilgenfeld R (2016) Science 7:503–505. https://doi.org/10.1126/science.aag2419
Xu M, Lee EM, Wen Z, Cheng Y, Huang W, Qian X, Kouznetsova J, Ogden SC, Hammack C, Jacob F, Nguyen HN, Itkin M, Hanna C, Shinn P, Allen C, Michael SG, Simeonov A, Huang W, Christian KM, Goate A, Brennand KJ, Huang R, Xia M, Ming G, Zheng W, Song H, Tang H (2016) Nat Med 22:1101–1107. https://doi.org/10.1038/nm.4184
Yuan S, Chan JF, den-Haan H, Chik KK, Zhang AJ, Chan CC, Poon VK, Yip CC, Mak WW, Zhu Z, Zou Z, Tee K, Cai J, Chan K, de la Pena J, Perez-Sanchez H, Ceron-Carrasco JP, Yuen K (2017) Antivir Res 145:33–43. https://doi.org/10.1016/j.antiviral.2017.07.007
Aleshin AE, Shiraev SA, Strongin AY, Liddington RC (2007) Protein Sci 16:795–806. https://doi.org/10.1110/ps.072753207
Noble CG, She CC, Chao AT, Shi PY (2012) J Virol 86:438–446. https://doi.org/10.1128/JVI.06225-11
Robin G, Chappell K, Stoermer MJ, Hu SH, Young PR, Fairlie DP, Martin JL (2009) J Mol Biol 385:1568–1577. https://doi.org/10.1016/j.jmb.2008.11.026
Chandramouli S, Joseph JS, Daudenarde S, Gatchalian J, Cornillez-Ty C, Kuhn P (2010) J Virol 84:3059–3067. https://doi.org/10.1128/JVI.02044-09
Hammamy MZ, Haase C, Hammami M, Hilgenfeld R, Steinmetzer T (2013) Chem Med Chem 8:231–241. https://doi.org/10.1002/cmdc.201200497
Assenberg R, Mastrangelo E, Walter TS, Verma A, Milani M, Owens RJ, Stuart DI, Grimes JM, Mancini EJ (2009) J Virol 83:12958–12906. https://doi.org/10.1128/JVI.00942-09
Phoo WW, Li Y, Zhang Z, Lee MY, Loh YR, Tan YB, Ng EY, Lescar J, Kang C, Luo D (2016) Nat Commun 7:13410. https://doi.org/10.1038/ncomms13410
Zhang Z, Li Y, Loh YR, Phoo WW, Hung A, Kang C, Luo D (2016) Science 354:1597–1600. https://doi.org/10.1126/science.aai9309
Lovell SC, Davis IW, Arendall III WB, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC (2003) Proteins 50:437–450. https://doi.org/10.1002/prot.10286
Trott O, Olson AJ (2010) J Comput Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Sterling T, Irwin JJ (2015) J Chem Inf Model 55:2324–2337. https://doi.org/10.1021/acs.jcim.5b00559
Baell JB, Walters MA (2014) Nature 513:481–483. https://doi.org/10.1038/513481a
Monga M, Sausville EA (2002) Leukemia 16:520–526. https://doi.org/10.1038/sj.leu.2402464
Li H, Leung K, Wong M (2015) CIBCB 34:115–126. https://doi.org/10.1109/CIBCB.2012.6217214
Butina D (1999) J Chem Inf Comput Sci 39:747–750. https://doi.org/10.1021/ci9803381
Ericksen SS, Wu H, Zhang H, Michael LA, Newton MA, Hoffman FM, Wildman SA (2017) J Chem Inf Model 57:1579–1590. https://doi.org/10.1021/acs.jcim.7b00153
Kukol A (2011) Eur J Med Chem 46:4661–4664. https://doi.org/10.1016/j.ejmech.2011.05.026
Case DA, Betz RM, Cerutti DS, Cheatham III TE, Darden TA, Duke RE, Giese TJ, Gohlke H et al (2016) AMBER. University of California, San Francisco
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE (2000) Acc Chem Res 33(12):889–897. https://doi.org/10.1021/ar000033j
Hou T, Wang J, Li Y, Wang W (2011) J Chem Inf Model 51(1):69–82. https://doi.org/10.1021/ci100275a
Genheden S, Ryde U (2015) Expert Opin Drug Discovery 10(5):449–461. https://doi.org/10.1517/17460441.2015.1032936
Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) J Chem Theory Comput 11(8):3696–3713. https://doi.org/10.1021/acs.jctc.5b00255
Wang J, Wang W, Kollman PA, Case DA (2004) J Comput Chem 25:1157–1174. https://doi.org/10.1002/jcc.20035
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) J Chem Phys 79:926–935. https://doi.org/10.1063/1.445869
Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC (2012) J Chem Theory Comput 8:1542–1555. https://doi.org/10.1021/ct200909j
Izaguirre JA, Catarello DP, Wozniak JM, Skeel RD (2001) J Chem Phys 114(5):2090–2098. https://doi.org/10.1063/1.1332996
Ryckaert J-P, Ciccotti G, Berendsen HJC (1977) J Comput Phys 23:327–341. https://doi.org/10.1016/0021-9991(77)90098-5
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) J Chem Phys 103(19):8577–8593. https://doi.org/10.1063/1.470117
Humphrey W, Dalke A, Schulten K (1996) J Mol Graph 14:27–38. https://doi.org/10.1016/0263-7855(96)00018-5
Gilson MK, Zhou HX (2007) Annu Rev Biophys Biomol Struct 36:21–42. https://doi.org/10.1146/annurev.biophys.36.040306.132550
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y (2012) J Chem Inf Model 52:3099–3105. https://doi.org/10.1021/ci300367a
Kim K, Shresta S (2016) Trends Microbiol 24:622–636. https://doi.org/10.1016/j.tim.2016.06.002
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 487 kb)
Rights and permissions
About this article

Cite this article
Bowen, L.R., Li, D.J., Nola, D.T. et al. Identification of potential Zika virus NS2B-NS3 protease inhibitors via docking, molecular dynamics and consensus scoring-based virtual screening. J Mol Model 25, 194 (2019). https://doi.org/10.1007/s00894-019-4076-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4076-6