
Abstract
To continue our previous work, the structure and some properties of a new series of 1,2,3,4-tetrazine 1,3-dioxides annulated with an imidazole ring or oxazole ring were studied in this paper. Four imidazolo-v-tetrazine 1,3-dioxides (ITDOs) I1–I4 and eight oxazolo-v-tetrazine 1,3-dioxides (OTDOs) O1–O8 were designed. We employed the density functional theory (DFT) in B3LYP/6-311++G(d,p) to study their geometrical structures and the homodesmotic reaction method to calculate the enthalpies of formation. Detonation properties and stabilities were also studied. Generally speaking, ITDOs and OTDOs have more preferable stabilities than TTDOs or pyrazolo-TDOs. I3, I4, O1, and O2 were found to be comparable to the energy level of RDX; O5 and O6 are even as powerful as HMX. The stabilities analysis in this paper can also prove that the five-membered ring deformation and the steric hindrance change caused by the different substituents will affect the stabilities of the structures of 1,2,3,4-tetrazine 1,3-dioxides annulated with a five-membered nitrogen-rich heterocycle. Other factors, such as the position of the electron-withdrawing substituents or the position of coordinated oxygen atom, are worthwhile to investigate in future work.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent decades, 1,2,3,4-tetrazine 1,3-dioxide (v-TDO), as a favorable structure unit, has become the choice for designing new high-energy-density compounds (HEDCs) [1,2,3,4,5,6,7,8], and many promising candidates have been obtained [9,10,11,12,13] based on its energetic skeleton. 5,7-Dinitrobenzo-1,2,3,4-tetrazine-1,3-dioxide (DNBTDO) has been studied by Klapötke et al. [11], and they found that the sensitivity of DNBTDO is slightly higher, while the detonation performance is slightly lower than that of RDX. [1,2,5]-oxadiazole-[3,4-e]-[1,2,3,4]-tetrazine-4,6-di-N-oxide (FTDO) [14,15,16] reduced the combustion temperature significantly and the signatures of the outlet gases when it was an additive of propellant compositions.
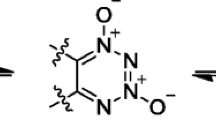
In our previous work, furoxano-v-tetrazine trioxide-β (FTTO-β) [17] was designed, and it is believed to be a considerable intermediate in the synthesis of monocyclic v-TDO. The structures mentioned above are shown in Fig. 1.

The structures of v-TDO, DNBTDO, FTDO, and FTTO-β
The v-TDO annulated with a pyrazole ring or 1,2,3-triazole ring, pyrazolo-v-TDOs, and 1,2,3-triazolo-TDOs (TTDOs), have been studied in our previous work [18, 19]. As shown in Table 1, some of them were found to have good detonation performances and acceptable stabilities, which compared with RDX. Their detonation performances and structures are listed in Table 1 and Fig. 2.

Structures of T2, T5, T11, P1, P4, and P11
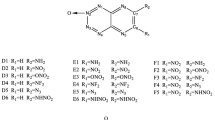
In this paper, v-TDO annulated with another five-membered ring was designed to continue our research. Four imidazolo-v-tetrazine 1,3-dioxides (ITDOs) I1–I4 and eight oxazolo-v-tetrazine 1,3-dioxides (OTDOs) O1–O8 were designed and shown in Fig. 3. Their properties were evaluated by the density functional theory (DFT). At the end of this paper, some points of view about design strategy for this kind of molecule are put forward after contrasting all of them, including FTDO, FTTOs, TTDOs, pyrazolo-TDOs, ITDOs, and OTDOs.

Designed ITDOs and OTDOs
Computational methods
The geometry optimization of the 12 target molecular structures was calculated by the B3LYP/6-311++G(d,p) level in the Gaussian software package [20]. At the same level, the vibrational analysis was performed, and the optimized structures were confirmed to be the local minimum without imaginary frequencies. According to the previous study, the calculated frequencies were scaled uniformly by 0.96 to approximately correct the systematic overestimation.








The homodesmotic reactions [21] designed as Eqs. (1–8) were employed to calculated the enthalpies of formation of I1–I4 and O1–O8 by Eq. (9):
where R or R’ is either NO2 or NH2; ΔH°298 is the change of enthalpy of formation at 298 K; ∑ΔfH°298, P and ∑ΔfH°298, R are the sum of the enthalpies of formation of products and the reactants, respectively; ΔE°298 represents the change of total energy at some temperature; Δ(pV) represents the change of the pressure p and volume V in product. ΔE°298 and Δ(pV) can be obtained by Eqs. (10) and (11), respectively.
where ΔΕ0, ΔΕΖPV, and ΔH°T are the change of total energy at 0 K, the zero-point vibrational energy, and the temperature-dependent enthalpy, respectively.
The solid-phase enthalpy of formation ΔfH°298(s) can be obtained by Eqs. (12) to (15):
where AS is the molecule surface area, σ+2 is the mean variance of the positive molecular electrostatic potential, and σ−2 is the negative one. The empirical parameters α1, α2, and α3 are obtained from [22]. The B3PW91/6-31G(d,p) level of DFT was employed to calculate the surface properties of sublimation enthalpies.
The densities (ρ) of these compounds can be obtained by Eq. (16) suggested by Politzer et al. [23]. The coefficients β1, β2, and β3 are 0.9183, 0.0028, and 0.0043, respectively:
where M is the molecular mass and V is the volume of the 0.001 electrons/bohr3 contour of the molecule electronic density.
The Kamlet-Jacobs equation is often employed to estimate the detonation velocity (D) and detonation pressure (P) of energetic materials [24,25,26].
Where N is the moles of gaseous products per gram explosives, \( \overline{M} \) is the products’ average molecule mass, and Q is the detonation heat. They are determined by the most exothermic principle and based on the most exothermic principle. The released energy can reach its maximum when the products are supposed to be only CO2, H2O, and N2.
In order to satisfy the demand of industry and military, insensitive high explosives are imperative. The sensitivity becomes an essential factor in design of energetic materials, and can be evaluated by many methods [27,28,29,30,31,32,33]. According to refs. [34,35,36,37,38,39], the solid compound’s compressibility and the crystal lattice free space (ΔV) are the factors related to sensitivity. According to Politzer’s findings, ΔV can be calculated as follows:
where Veff is the effective molecule volume when the cell unit was fully filled hypothetically. Vint is the intrinsic space encompassed by the 0.003 a.u. contour of the molecule’s electronic density. M is the molecular mass, and ρ is the crystal density.
The thermal stabilities of the ITDOs and OTDOs were further confirmed by the bond dissociation activation energy (BDAE) obtained by scanning the potential energy surface for pyrolysis.
Results and discussion
Molecular structures
The optimized structures of I1–I4 and O1–O8 are shown in Fig. 4 for comparison.

Molecular geometric parameters of ITDOs and OTDOs
Under the influence of hydrogen bond interactions, all the ITDOs and OTDOs are nonplanar structures; however, the OTDOs are quite close to fully planar ones. The absolute values of dihedral angles of O(5)-C(6)-N(12)-O(13), N(5)-C(6)-N(12)-O(13), O(5)-C(6)-N(12)-O(14), and N(5)-C(6)-N(13)-O(14) in O1, O2, O5, and O6 are less than 0.1 degree. The dihedral angles of N(5)-C(6)-N(12)-H(13) and N(5)-C(6)-N(13)-H(14) in O4 and O8 are 0.19166° and 0.11849°, respectively. The bond lengths in 12 compounds are all in the reasonable range, and we presume these imidazole and oxazole rings, like the v-TDO ring, are both solid enough.
Enthalpy of formation
In this paper, the enthalpies of formation of the 12 target compounds were calculated by the isodesmic reaction method and the designed homodesmotic reactions (1–8) were proposed properly.
The total energies E0 and enthalpies of formations ΔfH° of the small molecules in reactions (1–8) are listed in Table 2. Of these, ΔfH° values for 2-nitro-1H-imidazol-1-amine, 2-nitrooxazole, and oxazol-2-amine were calculated by G3 theory based on the atomization reaction CaHbOcNd(g) → aC(g) + bH(g) + cO(g) + dN(g).
Table 3 presents the corresponding parameters that are needed to calculate the solid-phase enthalpies of formation of I1–I4 and O1–O8. The results show that I4 has the highest ΔfH°298(s), while O3 has the lowest one.
Detonation properties
The calculated detonation properties according to Eqs. (16), (17), and (18) are listed in Table 4. The corresponding data of RDX and HMX are also provided. It shows that the detonation performances of O5 and O6 are as good as HMX. I3, I4, O1, and O2 can also achieve the RDX energy level.
Sensitivity
For most of the v-TDO compounds, according to the value of BDAEs calculations, the N–N bonds on pyrazole rings are above 200 kJ·mol−1, and the N=N bonds of pyrazine rings are above 280 kJ·mol−1. Affected by the substituent N–O, –NO2 or –NH2, the weak bonds of FTTOs [17], TTDOs [18], and pyrazolo-TDOs [19] mainly center on the five-membered rings. In addition, C–NO2 or N–NO2 may also be the trigger bonds for many nitro compounds. The BDAEs of some possible trigger bonds of 12 target compounds are shown in Table 5. The BDAEs of trigger bonds N–NO2 of RDX and HMX are also given in Table 5.
According to the research of Heming Xiao et al. [25], a BDAE or BDE of 80–120 kJ·mol−1 is usually considered to be an indicator for a stable energetic compound in HEDC molecular design. As shown in Table 5, the most fragile part of the imidazole rings in I1–I4 are bonds between nitro substituent and amino substituent or bonds between nitro substituent and coordinated oxygen atom, the BDAEs of which are 220.54, 194.29, 204.79, and 178.53 kJ·mol−1 respectively. BDAEs of the weakest bonds in nitro-substituented compounds O1, O2, O5, and O6 are near 200 kJ·mol−1, almost 50 kJ·mol−1 higher than the ones of amino-substituented compounds O3, O4, O7, and O8. The BDAEs of C–NO2 bonds in I1–I4 and O1, O2, O5, O6 are above 200 kJ·mol−1, which means they may not be the trigger bonds.
Sometimes using the BDAE or BDE values to measure the sensitivity is not accurate [33], and according to the research [39], the increase of sensitivity tends to follow the increase of the free space in the crystal lattices (ΔV). Table 5 lists the ΔV values of ITDOs and OTDOs, compared with RDX and HMX. It can be found that the OTDOs are more insensitive than the others. Of these, O5 (42.34 Å3) and O6 (42.71Å3) are more sensitive than O1 to O4. However, using the ΔV values to estimate sensitivity is a rough estimating method [41], so the sensitivities of ITDOs and OTDOs are roughly similar to RDX and HMX.
Conclusions
Generally speaking, a molecule of v-TDO annulated with a five-membered nitrogen-rich heterocycle owns good energy characteristics (Fig. 5). Compared to other molecules, ITDOs and OTDOs are no less energetic and are usually more stable. Some of them, such as I3, I4, O1, and O2, are promising materials as good as RDX; O5 and O6 are even as powerful as HMX.

Comparison of the Q (cal·g−1), D (m·s−1), and P (GPa) values of target v-TDOs and RDX
Compared with other v-TDOs, the v-TDO ring in this kind of compound is rather stable (Fig. 6) because the five-membered ring deformation and the steric hindrance change caused by the different substituents are the main reasons affecting the stabilities of these molecule structures. The more heteroatoms directly connected in the ring, the more obvious this effect performs. For this reason, the FTTOs and some designed TTDOs are quite unstable, compared to which the thermal stability of ITDOs, OTDOs, and the pyrazolo-TDOs are less affected. It is worth mentioning that FTDO and TTDOs without any electron-withdrawing substituents or atoms have good stability as well. N–NO2, which should always be given more attention than C–NO2, is an important potential trigger bond. For example, the average BDAE of the N–NO2 of T1, T2, P5, P6, P7, and P8 is about 110 kJ·mol−1, much lower than 280 kJ·mol−1, which is the average BDAE of the C–NO2 of other pyrazolo-TDOs, ITDOs, and OTDOs. Of course, we believe there are still other important factors to decide the stability of a v-TDO compound, for example, the position of the electron-withdrawing substituents or the position of coordinated oxygen atom, which may be worthwhile to investigate in future work.

Comparison of the BDAE of the weakest bond and the ΔV values of target v-TDOs and RDX
References
Tang Y, Yang H, Cheng G (2013) Synthesis and characterization of a stable, catenated N11 energetic salt. Angew Chem Int Ed 52:1–4
Wu Q, Zhu W, Xiao H (2014) A new design strategy for high-energy low-sensitivity explosives: combining oxygen balance equal to zero, a combination of nitro and amino groups, and N-oxide in one molecule of 1-amino-5-nitrotetrazole-3N-oxide. J Mater Chem A 2:13006–13015
Politzer P, Lane P, Murray JS (2013) Computational analysis of relative stabilities of polyazine N-oxides. Struct Chem 24:1965–1974
Politzer P, Lane P, Murray JS (2013) Tricyclic polyazine N-oxides as proposed energetic compounds. Cent Eur J Energ Mater 10:305–323
Politzer P, Lane P, Murray JS (2013) Computational characterization of two di-1,2,3,4-tetrazine tetraoxides, DTTO and iso-DTTO, as potential energetic compounds. Cent Eur J Energ Mater 10:37–52
Ye C, An Q, Goddard WA, Cheng T, Liu W, Zybin SV, Ju X (2015) Initial decomposition reaction of di-tetrazine-tetroxide (DTTO) from quantum molecular dynamics: implications for a promising energetic material. J Mater Chem A 3:1972–1978
Piercey DG, Chavez DE, Heimsch S, Kirst C, Klapötke TM, Stierstorfer J (2014) An energetic N-oxide and N-amino heterocycle and its transformation to 1,2,3,4-tetrazine-1-oxide. Propellants Explos Pyrotech. https://doi.org/10.1002/prep.201400224
Wang T, Zheng C, Yang J, Zhang X, Gong X, Xia M (2014) Theoretical studies on a new high energy density compound 6-amino-7-nitropyrazino[2,3-e][1,2,3,4]tetrazine 1,3,5-trioxide (ANPTTO). J Mol Model 20:2261–2271
Churakov AM, Tartakovsky VA (2004) Progress in 1,2,3,4-tetrazine chemistry. Chem Rev 104:2601–2616
Bi F, Wang B, Li X, Fan X, Cheng X, Ge Z (2012) Progress in the energetic materials based on 1,2,3,4-tetrazine 1,3-dioxide. Chin J Energ Mater 5:630–637
Klapötke TM, Piercey DG, Stierstorfer J, Weyrauther M (2012) The synthesis and energetic properties of 5,7-dinitrobenze-1,2,3,4-tetrazine-1,3-dioxide. Propellants Explos Pyrotech 37:527–535
Voronin AA, Zelenov VP, Churakov AM, Strelenko YA, Fedyanin IV, Tartakovsky VA (2014) Synthesis of 1,2,3,4-tetrazine 1,3-dioxides annulated with 1,2,3-triazoles and 1,2,3-triazole 1-oxides. Tetrahedron 70:3018–3022
Voronin AA, Zelenov VP, Churakov AM, Strelenko YA, Tartakovsky VA (2014) Alkylation of 1-hydroxy-1H-[1,2,3]triazolo[4,5-e][1,2,3,4]tetrazine 5,7-dioxide. Russ Chem Bull Int Ed 63:475–479
Churakov AM, Ioffe SL, Tarakovsky VA (1995) Synthesis of [1,2,5]oxadiazolo[3,4-e][1,2,3,4]tetrazine 4,6-di-N-oxide. Mendeleev Commun 5:227–228
Zelenov VP, Lobanova AA, Lyukshenko NI, Sysolyatin SV, Kalashnikov AI (2008) Behavior of [1,2,5]oxadiazolo[3,4-e][1,2,3,4]tetrazine 4,6-dioxide in various media. Russ Chem Bull Int Ed 57:1384–1389
Zelenov VP, Lobanova AA, Sysolyatin SV, Sevodina NV (2013) New syntheses of [1,2,5]oxadiazolo[3,4-e][1,2,3,4]tetrazine 4,6-dioxide. Russ J Org Chem 49:467–477
Wang T, Zhang T, Xu L, Wu X, Gong X, Xia M (2014) Theoretical studies on vicinal-tetrazine compounds: furoxano-1,2,3,4-tetrazine-1,3,5-trioxide (FTTO-α) and furoxano-1,2,3,4-tetrazine-1,3,7-trioxide (FTTO-β). J Mol Model 20:2516–2527
Wang T, Zheng C, Liu Y, Gong X, Xia M (2015) Theoretical studies of structure, stability and detonation properties of vicinal-tetrazine 1,3-dioxide annulated with a five-membered heterocycle. 1. Annulation with a triazole ring. J Mol Model 21:201–209. https://doi.org/10.1007/s00894-015-2748-4
Wang T, Zheng C, Gong X, Xia M (2015) Theoretical studies of structure, stability and detonation properties of vicinal-tetrazine 1,3-dioxide annulated with a five-membered heterocycle. 2. Annulation with a pyrazole ring. J Mol Model 21:269–277. https://doi.org/10.1007/s00894-015-2816-9
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven JT, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, revision B.05. Gaussian Inc, Wallingford
Wheeler SE, Houk KN, Schleyer PVR, Allen WD (2009) A hierarchy of homodesmotic reactions for thermochemistry. J Am Chem Soc 131:2547–2560
Politzer P, Ma Y, Lane P, Concha MC (2005) Computational prediction of standard gas, liquid, and solid-phase heats of formation and heats of vaporization and sublimation. Int J Quantum Chem 105:341–347
Politzer P, Martinez J, Murray JS, Concha MC, Toro-Labbe A (2009) An electrostatic interaction correction for improved crystal density prediction. Mol Phys 107:2095–2101
Kamlet M, Jacobs SJ (1968). Chem Phys 48:23
Xiao HM, Xu XJ, Qiu L (2008) Theoretical design of high energy density materials. Science Press, Beijing
Politzer P, Murray JS (2011) Some perspective on estimating detonation properties of C, H, N, O compounds. Cent Eur J Energ Mater 8:209–220
Politzer P, Murray JS (1996) Relationships between dissociation energies and electrostatic potentials of C-NO2 bonds: applications to impact sensitivities. J Mol Struct 376:419–424
Zhang C, Shu Y, Huang Y, Zhao X, Dong H (2005) Investigation of correlation between impact sensitivities and nitro group charges in nitro compounds. J Phys Chem B 109:8978–8982
Xiao H, Li Y (1995) Banding and electronic structures of metal azides - sensitivity and conductivity. Sci Chin Ser B 38:538–545
Politzer P, Murray JS, Concha MC (1998) C-H and C-NO2 dissociation energies in some azines and nitroazines. J Phys Chem A 102:6697–6701
Cao X, Xiang B, Zhang C (2012) Review on relationships between the molecular and crystal structure of explosives and their sensitivities. Chin J Energ Mater 5:643–649
Tan B, Long X, Peng R, Li H, Jin B, Chu S (2011) On the shock sensitivity of explosive compounds with small-scale gap test. J Phys Chem A 115:10610–10616
Tan B, Long X, Li J, Nie F (2012) Insight into shock-induced chemical reaction from the perspective of ring strain and rotation of chemical bonds. J Mol Model 18:5127–5132
Dlott DD (2003) Fast molecular aspects in energetic materials. In: Politzer P, Murray JS (eds) Energetic materials. Part 2. Detonation, combustion, vol 6. Elsevier, Amsterdam, pp 125–191
Tsai DH, Armstrong RW (1994) Defect-enhanced structural relaxation mechanism for the evolution of hot spots in rapidly compressed crystals. J Phys Chem 98:10997–11000
Kunz AB (1996) An ab initio investigation of crystalline PETN. Mater Res Soc Symp Proc 418:287–292
Lide DR (2009) CRC handbook of chemistry and physics, 90th edn. CRC Press, Boca Raton
Pospisil M, Vavra P, Concha MC, Murray JS, Politzer P (2011) Sensitivity and the available free space per molecule in the unit cell. J Mol Model 17:2569–2574
Politzer P, Murray JS (2015) Some molecular/crystalline factors that affect the sensitivities of energetic materials: molecular surface electrostatic potentials, lattice free space and maximum heat of detonation per unit volume. J Mol Model 21:25. https://doi.org/10.1007/s00894-015-2578-4
Rice BM, Mattson W, Trevino SF (1998) Molecular-dynamics investigation of the desensitization of detonable material. Phys Rev E 57:5106–5111
Tarver CM, Urtiew PA, Tran TD (2005) Sensitivity of 2,6-diamino-3,5-dinitropyrazine-1-oxide. J Energ Mater 23:183–203
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zheng, C., Wang, T., Wang, F. et al. Theoretical studies on a new series of 1,2,3,4-tetrazine 1,3-dioxide annulation with an imidazole ring or oxazole ring. J Mol Model 25, 36 (2019). https://doi.org/10.1007/s00894-018-3918-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-018-3918-y