
Abstract
Angiotensin-converting enzyme (ACE) converts angiotensin I to angiotensin II and degrades bradykinin and other vasoactive peptides. ACE inhibitors are used to treat diseases such as hypertension and heart failure. It is thus highly desirable to understand the catalytic mechanism of ACE, as this should facilitate the design of more powerful and selective ACE inhibitors. ACE exhibits two different active domains, the C-domain and the N-domain. In this work, we systematically investigated the inhibitor- and substrate-binding patterns in the N-domain of human ACE using a combined quantum mechanical and molecular mechanical approach. The hydrolysis of hippuryl–histidyl–leucine (HHL) as catalyzed by the N-domain of human somatic ACE was explored, and the effects of chloride ion on the overall reaction were also investigated. Two models, one with and one without a chloride ion at the first binding position, were then designed to examine the chloride dependence of inhibitor–substrate binding and the catalytic mechanism. Our calculations indicate that the hydrolysis reaction follows a stepwise general base/general acid catalysis path. The estimated mean free energy barrier height in the two models is about 15.6 kcal/mol, which agrees very well with the experimentally estimated value of 15.8 kcal/mol. Our simulations thus suggest that the N-domain is in a mixed form during ACE-catalyzed hydrolysis, with the single-chloride-ion and the double-chloride-ion forms existing simultaneously.

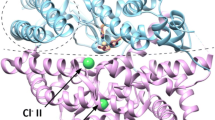
Superposition of ACE C- and N- domains
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Angiotensin I-converting enzyme (ACE, EC 3.4.15.1) is a zinc-containing dipeptidase that was discovered in 1956 [1]. It plays a key role in converting angiotensin I to angiotensin II, a powerful vasoconstrictor [2]. It can also degrade bradykinin, a potent vasodilator, and other vasoactive peptides [3]. The application of ACE inhibitors is considered to be a highly effective strategy for treating hypertension and cardiovascular diseases [4, 5]. The crystal structure of ACE was first reported in 2003, along with the structure of its complex with the inhibitor lisinopril [6], and the X-ray structures of other ACE inhibitors have since been resolved [6, 7].
Human somatic ACE (sACE) is a zinc-containing dipeptidase with two functional domains: the C-domain and the N-domain. These domains present an amino acid sequence similarity of 55 % [8]. The unique physiological roles of the N- and C-domains have been highlighted, along with the negative cooperativity between them [9, 10]. It was observed in domain-selective inhibition experiments that the C-domain plays the dominant physiological role in regulating hypertension, while the N-domain has little effect [11]. Moreover, inhibition of the N-domain can aid recovery from tissue injury and fibrotic disease without affecting blood pressure. The existence of two domains calls for the design of domain-selective inhibitors, since early-stage ACE inhibitors such as captopril, lisinopril, and enalaprilat show little domain selectivity and are thus associated with adverse side effects. Indeed, some domain-selective inhibitors have been reported, such as the two phosphinic peptide inhibitors RXP407 [11] (for the N-domain) and RXPA380 [12] (for the C-domain). It is worth noting that another form of ACE is found in the testes and plays a role in fertilization; this testicular ACE (tACE) [13] has nearly the same amino acid sequence in its C-domain as that in sACE, except for the first 36 residues.
The widespread usage of ACE inhibitors underscores the importance of understanding how the N- and C-domains participate in binding and catalysis at the microscopic level. Figure 1 shows the X-ray structure of the N-domain superposed on the X-ray structure of the C-domain. The C-domain (using tACE as the template [6]) and the N-domain [14] present nearly the same active-site conformation, which indicates that both domains utilize very similar catalytic mechanisms. However, some distinct differences between the domains in terms of inhibitor recognition can be observed. For example, T496 at the S1 subsite of the N-domain is the counterpart to V518 at the S1 subsite of the C-domain, indicating that there are different polarization environments at the S1 subsites of the two domains. In addition, a glutamate residue (E162) is present at the S1′ site in the C-domain, while no such residue is found in the N-domain. Indeed, it has been suggested that the interactions of inhibitors with residues unique to a particular domain is the mechanism for domain-specific inhibition [15]. However, this cannot explain the binding and catalytic specificities of the domains. Some highly domain-specific substrates have been reported. For example, the circulating tetrapepetide N-acetyl-SDKP [16] and angiotensin 1–7 (DRVYIHP) [17] are N-domain-specific substrates.

Panel a shows the N-domain (PDB code 2C6N, [14]; green) superposed on the C-domain (PDB code 1O86, [6]; cyan) of ACE complexed with lisinopril. The active sites and positions of chloride ions are highlighted in panel b (chloride ions in the C-domain are colored orange, while those in the N-domain are shown in green; all C-domain residue codes derive from tACE indexing)
Another important structural feature of ACE is that its catalytic activity is dependent on the presence of chloride ions. The degree of dependence was found to be substrate-specific [18–21]. In the C-domain of ACE, two chloride ions have been identified outside the active site, as shown in Fig. 1. More interestingly, experimental results suggest that these two chloride ions have different functions [15, 20, 22, 23]. The chloride ion at the second binding pocket, which is about 10 Å from the zinc ion, may influence catalytic activity via long-range electrostatic interactions [20, 23]. The chloride ion at the first binding position, which is more than 20 Å from the zinc ion, is thought to be important for maintaining structural stability [22]. However, in the X-ray structure of the N-domain (see Fig. 1), only one chloride ion was observed at the second binding pocket (hereafter termed Cl−(II)). It should be noted that experimental studies also found that the N-domain is activated at a lower chloride ion concentration and to a lesser degree than the C-domain [24, 25]. Thus, researchers suggested that the chloride ion at the first binding position (hereafter termed Cl−(I)) may not be necessary for the N-domain to be catalytically active [14]. A recent site-directed mutagenesis and kinetics study, on the other hand, suggested that current data do not provide sufficient evidence to clarify whether or not Cl−(I) occurs at its binding pocket [21]. In their study, the catalytic activities of the N-domain toward some substrates, e.g., hippuryl–histidyl–leucine (HHL) and Ang-I, were found to be even higher than than those of the C-domain at low chloride concentrations. However, we must point out that a low salt concentration does not imply the complete absence of Cl−(I) from the enzyme system. In fact, considering the high degree of similarity between the C- and N-domains of ACE and that both domains have nearly the same binding pocket for chloride ions, it would be very useful to determine the possibility of a chloride ion occurring at the first binding position in the N-domain, as well as the function of this chloride ion if it does.
We previously constructed a Michaelis complex model of the C-domain of sACE bound to HHL [26]. A general base/general acid (GAGB) or promoted-water mechanism was subsequently proposed for the hydrolysis of HHL catalyzed by ACE [27]. It was suggested that the zinc-bound water is a nucleophile that is activated by the residue E384 at the active site and polarized by a zinc ion. The zinc ion serves as an oxyanion hole to stabilize the tetrahedral intermediate (EI) complex. Finally, to complete the catalytic reaction, this hydrogen atom migrates to the amide nitrogen atom, breaking the C–N bond. It was reported that several peptidases utilize a similar mechanism to catalyze hydrolysis reactions [28]. A subsequent combined DFT/MM study of the C-domain of ACE confirmed this proposed mechanism [29]. More detailed information on the ACE N-domain is clearly required to aid the development of domain-selective inhibitors. Therefore, in the work reported in the present paper, we focused on the inhibitor–substrate binding mode and the corresponding catalytic mechanism for the N-domain. The chloride ion dependence was also addressed. We believe that our work provides a complete picture of the catalytic processes of the ACE.
Computational details
It is widely accepted that hybrid methods, for example combined quantum mechanical and molecular mechanical (QM/MM) methods, are suitable for electronic structure calculations of large systems such as enzymes [30]. Their fundamental principles and their applications to enzyme systems have been summarized in many peer-reviewed works [28, 31–34]. In the present study, a semi-empirical method—the self-consistent-charge density functional-tight binding (SCC-DFTB) method [35, 36]—was selected for our QM/MM simulations. Because parameters for biological zinc ions have been specifically developed for the SCC-DFTB method [37], we have applied this method to several zinc-containing enzymes, such as metallo-β-lactamases [38–40], aminopeptidases [41], and ACE [27]. It has been shown to give accurate results in both geometry and reaction mechanism simulations. The atoms in the MM region were simulated using the CHARMM all-atom force field [42].
Enzyme–substrate complex model
The initial structure for the N-domain of human sACE (bound to lisinopril) was extracted from the Protein Data Bank (PDB entry code 2C6N) [14]. The original crystal structure was a dimer. In this work, only chain A was considered in our simulations. According to Scheme 1, the HHL molecule has nearly the same backbone structure as lisinopril. It was therefore logical to construct the initial enzyme–substrate (ES) complex using lisinopril as the template, just as we did for the C-domain. The possible binding structure and atom labeling scheme used are shown in Scheme 1b. The labeling scheme used for the atoms of the protein was based on the CHARMM convention. Similar to its coordination in the C-domain–substrate complex, the zinc ion is tetracoordinated with H361, H365, E389, and an active-site water molecule. This water molecule is also hydrogen bonded to E362 and occupies the appropriate position for the subsequent nucleophilic attack (NA) that breaks the amide C–N bond of the substrate. Therefore, in the ES complex, E362 is in its negatively charged form. This initial structure also resembles the ES structures of CPA [41] and TLN [43, 44]. The tautomeric states of the histidine residues were determined by carefully examining the surrounding hydrogen-bond network. Other titratable residues were considered to be in their usual protonated states at neutral pH. Some disulfide bonds between C128 and C136, C330 and C348, and C516 and C528 were enforced.

Atom labeling scheme used and the interactions between lisinopril (A) or Hip-His-Leu (B) and active-site residues of the N-domain of ACE
The ACE–HHL complex was then solvated in a pre-equilibrated TIP3P water sphere [45] with a radius of 30 Å centered on the zinc ion. A stochastic boundary condition (SBC) [46] was applied to reduce computational costs. Atoms more than 30 Å from the origin were deleted, while atoms 27–30 Å away were subjected to Langevin dynamics; this region was defined as a buffer region. Atoms less than 27 Å from the origin were free to move in a manner governed by Newtonian mechanics. Hydrogen atoms were added using the HBUILD module in the CHARMM program. The link atom approach was applied to describe the covalent interface between the QM and MM regions, particularly the hydrogen atoms inserted between the alpha and beta carbons.
As shown in Scheme 1b, the QM region of the ES complex consisted of the whole substrate molecule of HHL, the zinc ion and its ligands (the H361, H365, and E389 side-chain groups and the zinc-bound water molecule), and the putative general base/acid catalyst E362. This comprised a total of 105 atoms for the QM region.
Since the Cl− ions were outside the active site, they were simulated in the MM region. A 5.4-ns MD simulation was performed with an integration time step of 1.0 fs. The temperature was slowly increased to 300 K in 30 ps, and a 370-ps MD simulation was performed for further equilibration. A subsequent 5-ns MD trajectory was used for data analysis. During the simulation, the SHAKE algorithm [47] was applied to retain all covalent bonds involving hydrogen atoms. A group-based switching scheme [48] was applied for nonbonded interactions with a cutoff of 13 Å.
Models of chloride ion at the first binding position
Another interesting issue relating to the structure of 2C6N is that only one chloride ion was found in the crystal structure, in contrast to the reported structure for the C-domain of ACE. Two chloride ions were noted outside the active site of the C-domain, and these two chloride ions were suggested to play different physiological roles, providing either structural stabilization or catalytic ability. Therefore, it seemed reasonable to insert an additional chloride ion into the N-domain at the first binding position. According to Fig. 1, we know that the chloride ion at the second position is located more than 10 Å from the zinc ion, which remains between Y202 and R500 of the N-domain (Y224 and R522 of the C-domain). In the C-domain, Cl−(I) stays about 21 Å away from the zinc ion, which is hydrogen bonded to R186, W485, and R489 and participates in hydrophobic interactions with W486 (note that the residue index of tACE is used for residue labeling in this work). Due to the identical position of Cl−(II) in the two domains, it is natural to envisage that, if the N-domain has two chloride ions outside the active site, Cl−(I) of the N-domain will have a similar recognition status to Cl−(I) of the C-domain. In this work, we built a model that contained two chloride ions outside the active site, with the Cl−(I) ion manually docked into the enzyme. Its putative Cartesian coordinates were set to (−12.321, −7.264, −37.416) with respect to the original atomic coordinates in the X-ray structure. The Cl−(I) ion occupied the cavity formed by the residues H164, W463, W464, and R467. When the Cl−(I) was inserted, H164 was in its positively charged form due to a strong hydrogen bond with the backbone amide oxygen atom of W160. H164 occurred in the N-domain at the position occupied by R186 in the C-domain, as shown in Fig. 1. Finally, MD and reaction simulations were performed with the same setup protocol for this double-chloride-ion model to examine the need for and the functional role of Cl−(I).
Potentials of mean force
MD simulations of the complex can only tell us the geometry of substrate binding; they cannot give us detailed information on reaction processes. Due to the almost identical active-site environments of the C- and N-domains of sACE, it should be straightforward to use the same computational strategy for the N-domain as already used for the C-domain. A proposed GAGB mechanism is given in Scheme 2; this mechanism is essentially the same as that for the C-domain of ACE and for other peptidases such as CPA and TLN. The reaction coordinate for the first step is defined as \( {r}_1={d}_{{\mathrm{C}}_{4\cdots }{\mathrm{O}}_{\mathrm{W}}} \) , which relates to the nucleophilic attack (NA) of the zinc-bound water at the amide carbon atom (C4) and the migration of a proton from the water molecule to E362. The reaction coordinate of the second elimination step (E) is then defined as \( {r}_2={d}_{{\mathrm{H}}_1\cdots {\mathrm{O}}_{\varepsilon_1}(E362)}-{d}_{{\mathrm{H}}_{1\cdots }{\mathrm{N}}_5} \), which involves the transfer of a proton from E362 to the amide nitrogen N5 and the cleavage of the amide bond. First, we calculated the minimum energy path (MEP) along the putative reaction coordinates using the adiabatic mapping approach (sometimes termed the coordinate driving method). To include the fluctuations and reorganization of the overall protein environment, we computed the potentials of mean force (PMFs) for the hydrolysis of the target peptide substrate. The umbrella sampling method was applied to enhance the sampling around the peak region with constraint force constants of about 100–300 kcal/(mol Å2). Eighteen and 21 windows were used for the NA and E steps, respectively. A total of 100 ps of constrained MD simulations were carried out for each window; the first 60 ps for heating and equilibration and the remaining 40 ps of the trajectory for the final PMF analysis. We also calculated multiple PMF profiles starting from the different initial ES structures extracted from the MD trajectory in order to include statistical contributions. Finally, the weighted histogram analysis method (WHAM) [49, 50] was applied to obtain the complete PMFs along the putative reaction coordinates. During the PMF simulations, the SHAKE module was applied to all hydrogen atoms except for the hydrogen atoms on the zinc-bound water molecule.

Proposed catalytic mechanism of the hydrolysis of hippuryl–histidyl–leucine as catalyzed by the N-domain of ACE
Results and discussion
Dynamics of the Michaelis complex
We first carried out molecular dynamics simulations to establish the binding modes of the N-domain of ACE complexed with lisinopril (further details of this can be found in the “Electronic supplementary material” (ESM). The calculated structure of the ACE/lisinopril complex was quite similar to its X-ray structure, which provided encouragement that the SCC-DFTB/CHARMM method could be used to investigate the enzymatic mechanism of ACE.
The best way to gain an accurate and deep understanding of the enzymatic mechanism would be to directly analyze the complex structure of the enzyme and its bona fide substrate. Unfortunately, in most cases, due to their highly efficient catalytic abilities, it is very hard to obtain the X-ray structures of enzyme–substrate complexes. Therefore, it seemed logical to construct a possible Michaelis complex based on the structure of the enzyme–inhibitor or enzyme–product complex. The crystal structure of the ACE–lisinopril complex represents an excellent starting point when attempting to construct the structure of the enzyme–substrate complex because lisinopril and HHL have similar backbones, as shown in Scheme 1. The HHL molecule was used by Cushman et al. as a template when designing ACE inhibitors [51]. Since human sACE contains two active domains, the N- and C-domains, it would be useful to determine any differences in the substrate binding features of the two domains. This could lead to new avenues to explore in the design of domain-selective ACE inhibitors.
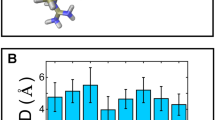
In this work, 5.4-ns MD simulations were carried out to examine the behavior of the constructed N-domain when complexed with the substrate in the absence (model I) and the presence (model II) of the Cl−(I) ion. As shown in Fig. 2, the geometry and structure of the systems remained largely the same throughout the MD simulations, with the backbone RMSDs calculated as 1.05 ± 0.10 Å (model I) and 1.20 ± 0.05 Å (model II), respectively. Selected key geometric data are summarized in Table 1 for both models. Snapshots of the two models extracted from the simulation trajectories are superposed in Fig. 3. They are almost identical, regardless of whether Cl−(I) exists. Clearly, the presence of Cl−(I) does not induce significant structural changes upon substrate binding, which is consistent with the experimental observation of the low chloride ion dependence of the N-domain [52].

Root mean square deviations (RMSDs) for models I and II of the enzyme–substrate complex as functions of time

Superposition of snapshots of the ES complex in the absence (pink) and the presence (yellow) of the Cl−(I) ion, as extracted from MD trajectories
As shown in Scheme 1b, in our Michaelis complex model, one zinc-bound water molecule and a neutral carbonyl group replaced the carboxylate group of lisinopril. In the MD model of the N-domain of ACE, the putative water molecule was found to remain at the active site throughout the simulation. The ligand bond that formed between the active-site water and the zinc ion remained largely intact throughout the simulation, given that \( {d}_{{\mathrm{O}}_{\mathrm{w}}\cdots \mathrm{Z}\mathrm{n}}=2.02\pm 0.06\kern0.5em \mathring{\mathrm{A}} \) for both models. At the same time, this water molecule was hydrogen bonded to the nearby glutamate residue E362, with bond distances of 1.21 ± 0.04 and 1.24 ± 0.05 Å for the two models. The occurrence of a similar binding motif for the zinc ion in several peptidases suggests that the downstream residue E362 acts as a general base in the first step, activating the zinc-bound water molecule. The hydrogen-bond network observed in the present work again verifies this suggestion, as it is very similar to the network we observed in simulations of the C-domain. Such hydrogen bonding and ligation with the zinc ion cause the water to occupy the perfect position for the subsequent nucleophilic attack reaction at the scissile carbonyl carbon, with \( {d}_{{\mathrm{O}}_{\mathrm{w}}\cdots {\mathrm{C}}_5}=3.29\pm 0.20\kern0.5em \mathrm{and}\kern0.5em 3.53\pm 0.35\mathring{A} \) noted for models I and II, respectively. Our model differs from that proposed by Sturrocks et al. [8], in which the amide carbonyl oxygen atom at the central recognition site interacts directly with the zinc ion. Instead, there was no direct contact between the substrate and the zinc ion in our simulations, as evidenced by \( {d}_{{\mathrm{O}}_2\cdots \mathrm{Z}\mathrm{n}}=4.04\pm 0.25\kern0.5em \mathring{A} \) for model I and 4.34 ± 0.40 Å for model II, respectively. This could indicate that the zinc ion can only play the role of a Lewis acid when activating the active-site water molecule. The scissile oxygen atom (O2) hydrogen bonds to the phenol group of Y501 with bond lengths of 1.74 ± 0.14 Å for model I and 2.14 ± 0.64 Å for model II. This interaction facilitates nucleophilic attack as it causes the polarization of the scissile carbonyl oxygen atom (O2), so Y501 acts as an oxyanion hole. It should also be noted that the geometric fluctuations in model II are somewhat larger than those in model I. These different binding patterns mean that the activity of the enzyme during the catalyzed hydrolysis of HHL may depend on the nature of the binding of the chloride ions. Discerning further effects on the reaction will require more investigations of substrate binding differences. Indeed, polarization of the carbonyl oxygen atom during hydrolysis occurs in several peptidases, such as Y248 in CPA [53] and R203 in TLN [54]. It was even thought to participate directly in the reaction catalyzed by CPA [55], although this was not supported by subsequent theoretical simulations [41]. Simulations carried out by Papakyriakou et al. [56] using the Amber force field suggest that the zinc ion is pentacoordinate. As a matter of fact, the pentacoordinate zinc configuration in TLN was found to be stable by Blumberger et al. when they performed calculations using the Amber force field [43], but the carbonyl oxygen was “expelled from the first coordinate shell” in the first picoseconds of their QM/MM simulation. Indeed, an ab initio/MM mechanistic simulation carried out by Ramos and coworkers [29] clearly supports the idea that the tetracoordinate model of the zinc ion is slightly more favorable than the pentacoordinate model. At the same time, because QM/MM MD was not employed in their simulation of substrate binding, their pentacoordinate model is questionable. The tetracoordinate model of the zinc ion therefore appears to be established and reasonable.
An extensive hydrogen network between HHL and various residues was observed in our simulations. This network stabilizes substrate binding. For example, the carboxylate group of the terminal Leu residue of the substrate forms strong hydrogen bonds with K489 and Y498. The central amide –NH group is partially stabilized by the backbone of A332, although some fluctuations were seen for both models. The backbone O7 atom is stabilized by H331 and H491 simultaneously in the absence of Cl−(I), but the hydrogen bond with H331 does not exist in the presence of Cl−(I). On the other hand, the electrostatic interactions between chloride ions and the enzyme environment are quite similar to those seen for the ACE–lisinopril complex.
Reaction pathway
The previous section detailed how we used the combined QM/MM method to construct Michaelis structures for the N-domain of ACE (complexed with the substrate of HHL) with or without Cl−(I). Starting from a structure randomly selected from the MD trajectory, we then performed reaction path calculations along the putative reaction coordinates using the adiabatic mapping approach (sometimes referred to as the reaction coordinate driving method; further details can be found in [57]). The resulting minimum energy paths were further refined using the conjugate peak refinement (CPR) approach [58]. It has been suggested that the CPR-corrected energy activation barrier height is a good approximation to the experimental value, especially when this approach is applied to proton-transfer reactions [59]. In the CPR approach, the reaction coordinate is then defined as
where N is the order of point i along the CPR trajectory, x(i) is the coordinate of point i, and n is the number of atoms. As also done by Friedman et al. [59], λ was normalized throughout this work (if not otherwise stated) such that 0 ≤ λ ≤ 1.
Our MD simulation of the substrate-binding mode suggested that the hydrolysis of HHL catalyzed by the N-domain of ACE proceeds via the general base/general acid (GAGB) mechanism, which is the same as that for the catalysis of HHL by the C-domain of ACE. Along with the envisaged GAGB reaction coordinates, the CPR-calculated reaction pathways are depicted in Fig. 4 for the two models. The geometric parameters for all five stationary states of model I are given in Table 2, while their topological representations are plotted in Fig. 5. All three minima of ES, the enzyme–intermediate complex (EI), and the enzyme–product complex (EP) were optimized using the adopted basis Newton–Raphson (ABNR) method [60, 61] with a stringent GMRS threshold of <0.0001 kcal/(mol Å2), and two transition states (TS1 and TS2) were optimized using the CPR module. We have included the CPR-refined geometric features and snapshots of the five stationary states for model II in the ESM, since they are similar to those for model I. We found that, no matter how many chloride ions were included in the calculations, the reaction mechanism was the same, with the first step being the rate-limiting step. The main change seen when the number of chloride ions was varied was the barrier height for each step. During the reaction, the zinc ion was generally coordinated to the protein residues. Some hydrogen bonds (as shown in Table 2) that were important for stabilizing the substrate also remained intact during the simulation. Significant geometric changes were only noted to occur around the reaction center.

CPR-refined energetic profiles obtained using the SCC-DFTB/MM approach for the hydrolysis of Hip–His–Leu by the N-domain of ACE with (line with black circles) and without (line with black triangles) the Cl−(I) ion

Stationary states for the hydrolysis of HHL catalyzed by the N-domain of ACE with only one chloride ion in the system (model I). The structures of the transition states were optimized using the conjugate peak refinement (CPR) approach
The hydrolysis of HHL catalyzed by ACE is a stepwise mechanism, with the first step involving the activation of the zinc-bound water molecule, the attack of the water molecule on the amide carbon (C4) atom, and the migration of a proton to E362. According to Table 2, as the water molecule (Ow) approaches the amide carbon (C4) atom, one of the hydrogen atoms (H1) migrates to the carboxylate group of E362; \( {d}_{{\mathrm{H}}_1\cdots {\mathrm{O}}_{\varepsilon 1}\left(\mathrm{E}362\right)} \) decreases from 1.20 to 1.01 Å in model I and from 1.18 to 1.04 Å in model II. This further confirms that E362 acts as a general base in this step. At the same time, the distance between the carbonyl oxygen atom and the zinc ion decreases sharply from 4.04 (ES) to 2.40 (TS1) Å in model I and from 3.39 Å (ES) to 2.32 Å (TS1) in model II. This behavior seems to be widespread in zinc-containing peptidases, with the zinc ion functioning as the oxyanion hole during the catalyzed reaction [28]. An almost pentacoordinate transition state (TS1) for the zinc ion can be observed in other peptidases such as CPA and TLN. With the formation of the intermediate, the fourth ligand of the zinc ion changes from the active-site water molecule to the carbonyl oxygen atom (O2), which results in a tetracoordinate zinc ion again. Although the hydrogen bond between O2 and Y501 was maintained throughout the reaction, the tetrahedral intermediate did not occur in our simulation if the fourth ligand did not form during the reaction. This highlights the function of the zinc ion from another perspective. As shown in Fig. 4, the CPR-refined energy barrier height was calculated to be 11.1 kcal/mol for model I and 14.7 kcal/mol for model II.
Once the tetrahedral intermediate is obtained, a shallow potential well (∼5.4 kcal/mol for model I and ∼4.5 kcal/mol for model II) must be overcome to complete the reaction (see Fig. 6). During this step, cleavage of the C4–N5 bond is facilitated by the migration of a proton from E362 to the amide nitrogen atom N5, with E362 functioning as a general acid. Finally, another hydrogen atom (H2) on the active-site water molecule transfers to E362 to neutralize this glutamate residue. This proton transfer resembles the binding of the inhibitor to the protonated side chain of E362 and a carboxylate group acting as the fourth ligand of the zinc ion. We have summarized the catalyzed reaction in Scheme 2.

Potential of mean force (PMF) for the hydrolysis of HHL catalyzed by the N-domain of ACE in the absence of the Cl−(I) ion. Free-energy profiles of different colors were calculated using different initial structures. Panel a refers to the first step (nucleophilic attack) while panel b refers to the second step (elimination)
As we emphasized in the section above, the original crystal structure of the N-domain of ACE contains only one chloride ion, which occupies the so-called second binding position. One of the major aims of this work was to determine the effects of the presence of a chloride ion at the first binding site. Based on our simulations, it is clear to see that the HHL molecule can be hydrolyzed by the N-domain of ACE in both chloride ion models. However, according to the CPR-optimized reaction pathway shown in Fig. 4, there are some distinct differences between the models, especially in terms of the barrier to the formation of the EI complex and the catalytic efficiency. Interestingly, the energy barrier height in model I is about 3.6 kcal/mol less than that in model II. In a previous simulation [27], we found that the enzymatic rate decreased when there was no chloride ion at the second binding site of the C-domain, due to the influence of long-range electrostatic interactions. For the N-domain of ACE, experimental results suggest that a low chloride ion concentration may facilitate catalyzed hydrolysis. The results of our current work agree with this experimentally derived conclusion, as the two models clearly have different catalytic abilities. However, since entropy contributions were not considered in the reaction path calculations, further free-energy profiles are required to verify this conclusion.
Calculated potential of mean force (PMF) profiles are depicted in Fig. 6 for model I and Fig. 7 for model II, respectively. To test the effects of different initial structures on the PMF, we randomly selected three ES structures from the MD trajectory for each model system. It is apparent that the overall shape of the free energy profile is similar to that obtained from our reaction path calculations, although the barrier height differs. Basically, the hydrolysis of HHL catalyzed by the N-domain of ACE is a stepwise reaction that involves nucleophilic attack at the scissile carbonyl carbon to form a tetrahedral intermediate and then proton migration to cleave the amide CN bond. For model I, in which there is only one chloride ion (occupying the second binding site), the activation barrier height for the first step of NA is calculated to be 13.9 ± 0.8 kcal/mol, while it is about 1.7 ± 0.3 kcal/mol for the second step (elimination). For model II, we included two chloride ions in the system. In particular, the putative chloride ion at the first binding site was inserted to examine its contribution to the overall catalyzed reaction. The calculated activation barrier height for the first step was calculated to be as high as 17.3 ± 0.8 kcal/mol and about 2.1 ± 0.5 kcal/mol for the second step. Current kinetic data indicate that the activation energy of the N-domain with respect to HHL is about 15.8 kcal/mol, as estimated from the experimental rate constant [21] of 14.15 ± 0.53 s−1 using transition state theory. Impressively, the activation energies in both models still show some deviation from the corresponding experimental value. However, the mean activation energy of the two models for the hydrolysis of HHL by the N-domain can be estimated to be ∼15.6 kcal/mol, in excellent agreement with the experimental kinetic data. Therefore, we suggest that the N-domain actually exists in a mixed form, i.e., the single-chloride-ion and the double-chloride-ion forms exist simultaneously. If this conclusion is true, then our results partially support the experimental observation that the N-domain shows a smaller chloride ion concentration dependence than the C-domain in the hydrolysis of peptide substrate molecules. At the same time, our simulations also explain why the X-ray structure with only one chloride ion can be crystallized. Merz and coworker [62] suggested that the QM/MM method should be applicable when attempting to refine the zinc-containing active-site structures of enzymes with low-resolution crystal structures. In this work, our results further indicate that combining MD with free-energy simulation using a QM/MM method could be useful for identifying some structural issues that occur outside the active site.

Potential of mean force (PMF) for the hydrolysis of HHL catalyzed by the N-domain of ACE in the presence of the Cl−(I) ion. Panel a refers to the first step (nucleophilic attack), while panel b refers to the second step (elimination)
On the other hand, according to the geometric parameters shown in Table 2, the insertion of Cl−(I) rarely affects the interactions between Cl−(II) and the protein environment, but it does cause some distinct structural fluctuations in the hydrogen-bond network that forms between the substrate and enzyme. This undoubtedly leads to decreased enzymatic activity, which is consistent with our PMF calculations. However, it should be noted that the helices that contain major residues which bind Cl−(I) are far from the active site. Therefore, the impact of the presence of Cl−(I) on the active site does not derive from direct electrostatic interactions. Nevertheless, interactions with the polar residues surrounding Cl−(I), as well as other residues, do influence the catalytic rate, although we cannot estimate the extent of this influence. Indeed, the importance of the binding site of Cl−(I) in the N-domain was recognized in previous works [15, 20], in which it was even suggested that residues at this site could inhibit enzymatic activity. We would expect substantial mutagenesis studies to address this issue, since it may be the key to understanding N- or C-domain-selective binding and the catalytic mechanism.
Finally, it is interesting to discuss domain selectivity based on our current work. Theoretical work that aims to elucidate the domain specificity of ACE is scarce. Combining our computational studies of the substrate binding and hydrolysis of both the C- and N-domains can yield some insight into this issue. Compared with Ang I or bradykinin, the HHL used in this work is much shorter. Because the N- and C-domains present a sequence similarity of ∼55 %, the zinc binding motif, active-site residues, and even the binding environment of the chloride ions are conserved between domains. More importantly, the differences between the two domains in the residues at the S1 and S1′ subsites do not lead to significant binding differences for two domains according to our simulations. In our simulations, in both domains, HHL did not interact directly with the zinc ion but it did form an extensive hydrogen-bond network with the protein. In addition, no matter whether experimental kinetic data or the results of our simulations were considered, the two domains were found to hydrolyze HHL with comparable catalytic efficiencies. Therefore, we can postulate that domain selectivity may not arise through variations in the arrangement of the active residues. A recent MD simulation study [48] of the recognition of gonadotropin-releasing hormone (GnRH) by the C- and N-domains of ACE suggested that it may interact directly with Cl−(II), which highlights the importance of chloride ion in substrate binding and enzymatic activity. It seems that the interactions between the substrate and the chloride ion and its surrounding residues may have an important role to play in domain specificity. More substantial studies of this topic are needed. Simulations of longer peptide substrates that focus on ACE domain selectivity would be highly desirable.
Conclusions
The physiological functions of ACE in regulating blood pressure and renal homeostasis make it an important drug target in the treatment of hypertension and other cardiovascular diseases. At the same time, it is also complicated and difficult to design ACE-domain-selective inhibitors because these two catalytically active domains are almost identical and they present an intrinsic chloride ion dependence. Only a few domain-selective inhibitors have been reported so far. In our previous simulations of the C-domain of ACE, we found that the chloride ion at the second binding site appears to make a positive contribution to the overall enzymatic activity via long-range electrostatic interactions, not by an allosteric mechanism. In the present work, we focused on the N-domain of human somatic ACE. Our interest was mainly in the chloride ion at the first binding site, Cl−(I). Using SCC-DFTB/CHARMM MD simulations, we systematically investigated the dynamic performance of a substrate (HHL) bound to the active site. We found that there are minor geometric differences between models with and without Cl−(I) at its putative binding site. Free-energy profiles calculated at the SCC-DFTB/MM level of theory showed that the overall hydrolysis reaction proceeds via the promoted-water or GAGB mechanism, in common with almost all zinc-containing peptidases. The rate-limiting step was shown to be the first step (nucleophilic attack) in both models. However, the system with only one chloride ion showed higher enzymatic activity, consistent with experimental observations [15] suggesting that the chloride ion at the first binding site and its surrounding residues may inhibit the catalytic mechanism to some degree. Moreover, the estimated mean active energy for hydrolysis was about 15.6 kcal/mol, in excellent agreement with the results obtained from experimental kinetic data, 15.8 kcal/mol. In other words, our simulations suggest that the N-domain of ACE is actually in a mixed form, with the single-chloride-ion N-domain and the double-chloride-ion N-domain existing simultaneously. It should be pointed out that the chloride ion dependence of the activity of ACE is highly dependent on the substrate employed. The substrate used in this work, HHL, is relatively short and cannot block the binding channel in ACE completely. To correctly reveal domain-selective mechanism, a longer substrate instead of HHL would be desirable. It is our hope that these results will stimulate more investigations aiming at a greater understanding of the ACE-domain-selective mechanism.
References
Skeggs LTJ, Kahn JR, Shuway NP (1956) The preparation and function of the hypertension-converting enzyme. J Exp Med 103:295–299
Ng KKF, Vane JR (1967) Conversion of angiotensin I to angiotensin II. Nature 216:762–766
Imig JD (2004) ACE inhibition and bradykinin-mediated renal vascular responses: EDHF involvement. Hypertension 43:533–535
Brown NJ, Vaughan DE (1998) Angiotensin-converting enzyme inhibitors. Circulation 97:1411–1420
Zaman MA, Poparil S, Calhoun DA (2002) Drugs targeting the renin–angiotensin–aldosterone system. Nat Rev Drug Discov 1:621–636
Natesh R, Schwager SLU, Sturrock ED, Acharya KR (2003) Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature 421:551–554
Natesh R, Schwager SLU, Evans HR, Sturrock ED, Acharya KR (2004) Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme. Biochemistry 43:8718–8724
Sturrock ED, Natesh R, van Rooyen JM, Acharya KR (2004) Structure of angiotensin I-converting enzyme. Cell Mol Life Sci 61:2677–2686
Binevski PV, Sizova EA, Pozdnev VF, Kost OA (2003) Evidence for the negative cooperativity of the two active sites within bovine somatic angiotensin-converting enzyme. FEBS Letters 550:84–88
Woodman ZL, Schwager SLU, Redelinghuys P, Carmona AK, Ehlers MRW, Sturrock ED (2005) The N domain of somatic angiotensin converting enzyme negatively regulates ectodomain shedding and catalytic activity. Biochem J 389:739–744
Junot C, Gonzales MF, Ezan E, Cotton J, Vazeux G, Michaud A, Azizi M, Vassiliou S, Yiotakis A, Corvol P, Dive V (2001) RXP 407, a selective inhibitor of the N-domain of angiotensin I-converting enzyme, blocks in vivo the degradation of hemoregulatory peptide acetyl-Ser-Asp-Lys-Pro with no effect on angiotensin I hydrolysis. J Pharmacol Exp Ther 297(2):606–611
Corradi HR, Chitapi I, Sewell T, Georgiadis D, Dive V, Sturrock ED, Acharya KR (2007) The structure of testis angiotensin-converting enzyme in complex with the C domain-specific inhibitor RXPA380. Biochemistry 46:5473–5478
Ehlers MRW, Fox EA, Strydom DJ, Riordan JF (1989) Molecular cloning of human testicular angiotensin-converting enzyme: the testis isozyme is identical to the C-terminal half of endothelial angiotensin-converting enzyme. Proc Natl Acad Sci USA 86:7741–7745
Corradi HR, Schwager SLU, Nchinda AT, Sturrock ED, Acharya KR (2006) Crystal structure of the N domain of human somatic angiotensin I-converting enzyme provides a structural basis for domain-specific inhibitor design. J Mol Biol 357:964–974
Moiseeva NA, Binevski PV, Baskin II, Palyulin VA, Kost OA (2005) Role of two chloride-binding sites in functioning of testicular angiotensin-converting enzyme. Biochem Mosc 70:1167–1172
Rousseau A, Michaud A, Chauvet MT, Lenfant M, Corvol P (1995) The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J Biol Chem 270:3656–3661
Deddish PA, Marcic B, Jackman HL, Wangh HZ, Skidgel RA, Erdos EG (1998) N-domain-specific substrate and C-domain inhibitors of angiotensin-converting enzyme. Hypertension 31:912–917
Cheung H-S, Wang F-L, Ondetti MA, Sabo EF, Cushman DW (1980) Binding of peptide substrates and inhibitors of angiotensin-converting enzyme. J Biol Chem 255:401–407
Shapiro R, Holmquist B, Riordan JF (1983) Anion activation of angiotensin converting enzyme: dependence on nature of substrate. Biochemistry 22:3850–3857
Liu X, Fernandez A, Wouters MA, Heyberger S, Husain A (2001) Arg(1098) is critical for the chloride dependence of human angiotensin I-converting C-domain catalytic activity. J Biol Chem 276:33518–33525
Yates CJ, Masuyer G, Schwager SLU, Akif M, Sturrock ED, Acharya KR (2014) Molecular and thermodynamics mechanisms of the chloride-dependent human angiotensin-I-converting enzyme (ACE). J Biol Chem 289:1798–1814
Bunning P, Riordan JF (1983) Activation of angiotensin conveting enzyme by monovalent anions. Biochemistry 22:110–116
Tzakos AG, Galanis AS, Spyroulias GA, Cordopatis P, Manessi-Zoupa E, Gerothanassis IP (2003) Structure-function discrimination of the N- and C- catalytic domains for human angiotensin-converting enzyme: implications from Cl− activation and peptide hydrolysis mechanism. Protein Eng 16:993–1003
Wei L, Alhenc-Gelas F, Corvol P, Clauser E (1991) The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J Biol Chem 266:9002–9008
Jaspard E, Wei L, Alhenc-Gelas F (1993) Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). J Biol Chem 268:9496–9503
Wang X, Wu S, Xu D, Xie D, Guo H (2011) Inhibitor and substrate binding by angiotensin-converting enzyme: quantum mechanical/molecular mechanical molecular dynamics studies. J Chem Inf Model 51:1074–1082
Zhang C, Wu S, Xu D (2013) Catalytic mechanism of angiotensin converting enzyme and effects of the choloride ion. J Phys Chem B 117:6635–6645
Xu D, Cui Q, Guo H (2014) Quantum mechanical/molecular mechanical studies of zinc hydrolysis. Int Rev Phys Chem 33:1–41
Brás NF, Fernandes PA, Ramos MJ (2014) QM/MM study and MD simulation on the hypertension regulator angiotensin-converting enzyme. ACS Catal 4:2487–2497
Warshel A, Levitt M (1976) Theoretical studies of enzymatic reactions: dielectric, electrostatic and steric stabilization of carbonium ion in the reaction of lysozyme. J Mol Biol 103:227–249
Gao J (1996) Methods and applications of combined quantum mechanical and molecular mechanical potentials. In: Lipkowitz KB, Boyd DB (eds) Reviews in computational chemistry, vol 7. VCH, New York, pp 119–185
Lin H, Truhlar DG (2007) QM/MM: what have we learned, where are we, and where do we go from here? Theor Chem Accounts 117:185–199
Senn HM, Thiel W (2009) QM/MM methods for biomolecular systems. Angew Chem Int Ed 48(7):1198–1229. doi:10.1002/anie.200802019
van der Kamp MW, Muloholland AJ (2013) Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 52:2708–2728
Cui Q, Elstner M, Kaxiras E, Frauenheim T, Karplus M (2001) A QM/MM implementation of the self consistent charge density functional tight binding (SCC-DFTB) method. J Phys Chem B 105:569–585
Elstner M (2006) The SCC-DFTB method and its application to biological systems. Theor Chem Accounts 116:316–325
Elstner M, Cui Q, Munih P, Kaxiras E, Frauenheim T, Karplus M (2003) Modeling zinc in biomolecules with the self consistent charge density functional tight binding (SCC-DFTB) method: applications to structure and energetic analysis. J Comput Chem 24:565
Xu C, Xie D, Zhang DH, Lin SY, Guo H (2005) A new ab initio potential energy surface of HO2(X2A′) and quantum studies of HO2 vibrational spectrum and rate constants for the H + O2 <−> O + OH reactions. J Chem Phys 122:244305
Xu D, Xie D, Guo H (2006) Catalytic mechanism of class B2 metallo-β-lactamase. J Biol Chem 281:8740–8747
Xu D, Guo H, Cui Q (2007) Antibiotic deactivation by a dizinc lactamase: mechanistic insights from QM/MM and DFT studies. J Am Chem Soc 129(35):10814–10822
Xu D, Guo H (2009) Quantum mechanical/molecular mechanical and density functional theory studies of a prototypical zinc peptidase (carboxypeptidase A) suggest a general acid-general base mechanism. J Am Chem Soc 131:9780–9788
MacKerell AD Jr, Bashford D, Bellott M, Dunbrack RL Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B102:3586–3616
Blumberger J, Lamoureux G, Klein ML (2007) Peptide hydrolysis in thermolysin; ab initio QM/MM investigation of the Glu143-assisted water addition mechanism. J Chem Theory Comput 3(5):1837–1850. doi:10.1021/ct7000792
Wu RB, Hu P, Wang SL, Cao ZX, Zhang YK (2010) Flexibility of catalytic zinc coordination in thermolysin and HDAC8: a Born–Oppenheimer ab initio QM/MM molecular dynamics study. J Chem Theory Comput 6(1):337–343. doi:10.1021/ct9005322
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Brooks CL III, Karplus M (1989) Solvent effects on protein motion and protein effects on solvent motion. J Mol Biol 208:159–181
Ryckaert J-P, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341
Steinbach PJ, Brooks BR (1994) New spherical-cutoff methods for long-range forces in macromolecular simulations. J Comput Chem 15:667
Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM (1992) The weighted histogram analysis method for free energy calculations on biomolecules. 1. The method. J Comput Chem 13:1011–1021
Roux B (1995) The calculation of the potential of mean force using computer simulations. Comput Phys Commun 91:275–282
Cushman DW, Cheung HS, Sabo EF, Ondetti MA (1977) Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry 16:5484–5491
Wei L, Clauser E, Alhenc-Gelas F, Corvol P (1992) The two homologous domains of human angiotensin I-converting enzyme interact differently with competitive inhibitors. J Biol Chem 267:13389–13405
Kilshtain-Vardi A, Glick M, Greenblatt HM, Goldblum A, Shoham G (2003) Refined structure of bovine carboxypeptidase A at 1.25 Å resolution. Acta Cryst D59:323–333
Gaucher JF, Selkti M, Tiraboschi G, Prange T, Roques BP, Tomas A, Fournie-Zaluski MC (1999) Crystal structures of α-mercaptoacyldipeptides in the thermolysin active site: structural parameters for a Zn monodentation or bidentation in metalloendopeptidases. Biochemistry 38:12569–12576
Cho JH, Kim DH, Kim D-H, Lee KJ, Choi KY (2001) The role of Tyr248 Probed by mutant bovine carboxypeptidase A: insight into the catalytic mechanism of carboxypeptidase A. Biochemistry 40:10197–10203
Papakyriakou A, Spyroulias GA, Sturrock ED, Manessi-Zoupa E, Cordopatis P (2007) Simulated interactions between angiotensin-converting enzyme and substrate gonadotropin-releasing hormone: novel insights into domain selectivity. Biochemistry 46:8753–8765
Woodcock HL, Hodoscek M, Brooks BR (2007) Exploring SCC-DFTB paths for mapping QM/MM reaction mechanisms. J Phys Chem A 111(26):5720–5728. doi:10.1021/jp0714217
Fischer S, Karplus M (1992) Conjugate peak refinement: an algorithm for finding reaction paths and accurate transition state in systems with many degrees of freedom. Chem Phys Lett 194:252–261
Friedman R, Fischer S, Nachliel E, Scheiner S, Gutman M (2007) Minimum energy pathways for proton transfer between adjacent sites exposed to water. J Phys Chem B 111:6059–6070
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) Charmm: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4:187–217
Chu JW, Trout BL, Brooks BR (2003) A super-linear minimization scheme for the nudged elastic band method. J Chem Phys 119:12708–12717
Li X, Hayik SA, Merz Jr KM (2010) QM/MM X-ray refinement of Zinc metalloenzymes. J Inorg Biochem 104:512–522
Acknowledgments
This work was funded by the National Natural Science Foundation of China (nos. 31170675 and 21473117). Some of the results described in this paper were obtained on the Deepcomp7000 at the Supercomputing Center of the Computer Network Information Center of the Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
Xia Mu and Chunchun Zhang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Computational details for the enzyme–inhibitor model. Results of the MD simulation of the complex between the N-domain of ACE and lisinopril are shown. Selected geometric parameters and corresponding snapshots of stationary states along the reaction coordinates for the N-domain with two chloride ions (model II) were calculated using the SCC-DFTB/MM/CPR framework. (DOCX 1966 kb).
Rights and permissions
About this article

Cite this article
Mu, X., Zhang, C. & Xu, D. QM/MM investigation of the catalytic mechanism of angiotensin-converting enzyme. J Mol Model 22, 132 (2016). https://doi.org/10.1007/s00894-016-3004-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-016-3004-2