
Abstract
Hemochromatosis is a clinical syndrome characterized by iron overload in various organs. We present here a case of type 4 hereditary hemochromatosis due to heterozygous mutation in SLC40A1 gene (p.D157A). SLC40A1 encodes ferroportin, a macromolecule only known as iron exporter from mammalian cells. He first presented symptoms correlated with hypopituitarism. Furthermore, marked hyperferritinemia and high transferrin saturation were revealed in combination with the findings of iron overload in the liver, spleen and pituitary gland by computed tomography and magnetic resonance imaging. Liver biopsy revealed iron deposition in both hepatocytes and Kupffer cells. SLC40A1 mutations are considered to cause wide heterogeneity by various ferroportin mutations. Thus, clinicopathological examinations seem to be very important for diagnosing phenotype of type 4 hemochromatosis in addition to the gene analysis. We diagnosed him as type 4B hereditary hemochromatosis (ferroportin-associated hemochromatosis) by the findings of high transferrin saturation and iron deposition in hepatocytes, and then started iron chelating treatment. We should suspect the possibility of hereditary hemochromatosis even in Japanese with severe iron overload. Although the same mutation in SLC40A1 gene (p.D157A) had been reported to cause “loss of function” phenotype, we considered that the mutation of our case caused “gain of function” phenotype.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hemochromatosis is a clinical syndrome characterized by iron accumulation and tissue injury in various organs presenting with complications such as chronic liver diseases (liver cirrhosis and hepatocellular carcinoma), diabetes mellitus, cardiac diseases, hypogonadotropic hypogonadism and endocrine dysfunction [1, 2]. Mutations in hereditary hemochromatosis-related genes are HFE, HJV, HAMP, TfR2 and SLC40A1. HFE-related hemochromatosis is most common in European populations, while the other mutations have been mainly reported in Asia-Pacific region [3, 4].
Mutations in SLC40A1, the gene encoding ferroportin, associate with an inborn error in iron metabolism transmitted through autosomal dominant inheritance, type 4 hereditary hemochromatosis [5, 6]. Ferroportin is the only macromolecule known to be able to release elemental iron from the mammalian cells and expressed in duodenal enterocytes, hepatocytes, Kupffer cells, splenic macrophages and erythroblasts [7]. Membrane expression of ferroportin is modulated post-translationally by hepcidin, a 25 amino acids protein, which binds to ferroportin and induces internalization and lysosomal degradation of ferroportin [8, 9]. Ferroportin activity controls the total iron content by transferring dietary iron from the enterocytes to plasma and also exports recycled and stored iron into the circulation from the hepatocytes and macrophages [10]. Although rare, type 4 hereditary hemochromatosis is observed in different groups of ethnic and is considered to be the second most common after HFE-related hemochromatosis [11].
In Japan, hereditary hemochromatosis is rare. We here describe a type 4B hereditary hemochromatosis Japanese patient first presenting with liver dysfunction and adrenal insufficiency. Furthermore, we demonstrate his heterozygous SLC40A1 gene mutation (p.D157A) which seemed to cause “gain of function” of ferroportin.
Case report
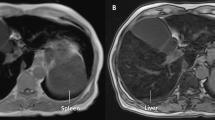
A 79-year-old man with disturbance of consciousness, general fatigue, appetite loss and vomiting admitted to local hospital. He was habitual drinker, about 20 g/day of alcohol consumption, and he had never taken supplemental iron and blood transfusion. He had a family history of liver cirrhosis in two brothers, and one of them was complicated with hepatocellular carcinoma. His son had intellectual disability. At the local hospital, laboratory test revealed liver dysfunction, hyponatremia (112 mmol/L), and low adrenocorticotropic hormone (ACTH, 5.3 pg/mL) and low cortisol (0.6 μg/dL). He was diagnosed with adrenal insufficiency, and then the local doctor started treatment with hydrocortisone (10 mg/day). After administration of hydrocortisone, appetite and general condition improved. Head and abdominal computed tomography (CT) demonstrated high density of pituitary gland (Fig. 1a, b), liver and spleen (Fig. 1c). Head and abdominal magnetic resonance imaging (MRI) demonstrated very low intensity of pituitary gland (Fig. 1d, e), liver and spleen (Fig. 1f) in T2-weighted image. Enlargement of the pituitary gland was also shown. He was referred to our hospital for evaluating cause of the liver dysfunction and adrenal insufficiency. On admission, physical examination showed no skin pigmentation. The results of laboratory examinations are shown in Table 1. Complete blood count revealed an anemia, while white blood cell and platelet count was within normal range. Biochemical examinations showed elevation of aspartate transaminase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP) and γ-glutamyltransferase (γ-GTP). The liver fibrosis markers, including hyaluronic acid, type IV collagen, Mac-2 binding protein glycosylation isomer (M2BPGi) and autotaxin, were elevated. Serum ferritin was significantly high. Total iron binding capacity (TIBC) and unsaturated iron binding capacity (UIBC) were low, and transferrin saturation was 95.8%. Fasting plasma glucose (FPG), HbA1c, serum insulin and C-peptide were within normal range. Homeostasis model assessment of insulin resistance (HOMA-IR) was 1.49. 75 g and oral glucose tolerance test was within normal range (data not shown). Hormonal examination revealed very low levels of serum thyroid-stimulating hormone (TSH) and free T4. Plasma luteinizing hormone (LH), estradiol and testosterone levels were low. Serum growth hormone (GH) level was within normal range, while insulin-like growth factor-1 (IGF-1) level was very low. Serum prolactin (PRL) level was elevated. Serum antidiuretic hormone (ADH) level and serum osmolarity were within normal range. Anterior pituitary function was evaluated by intravenous injection of corticotropin-releasing hormone (CRH), luteinizing hormone-releasing hormone (LHRH) and growth hormone-releasing peptide 2 (GHRP2) (Table 2). Plasma ACTH and cortisol showed low response to CRH. Plasma LH and follicle-stimulating hormone (FSH) also showed low response to LHRH. GH showed low response to GHRP2. Thus, he was diagnosed with hypopituitarism and secondary to hypothyroidism and hypogonadism. Electrocardiogram (ECG) and echocardiography were not remarkable change. Abdominal ultrasound showed liver cirrhosis pattern and moderate splenomegaly (not shown). Vibration-controlled transient elastography (Fibroscan®) showed 55.4 kPa [interquartile range (IQR); 10.6, IQR/median; 19%]. Esophagogastroduodenoscopy did not show esophageal gastric varices (not shown). Because hemochromatosis was suspected by the results of laboratory examinations and the findings of CT and MRI, liver biopsy was performed. Hematoxylin and eosin staining of the liver biopsy specimen showed chronic inflammatory infiltrate and periportal fibrosis without chronic non-suppurative destructive cholangitis (Fig. 2a, b, c). Berlin blue staining demonstrated iron accumulation in both hepatocytes and sinusoidal lining cells (probably Kupffer cells, arrowheads) (Fig. 2d). The hepatic iron concentration was 36,585 μg/g dry weight. Immunohistochemical findings of the liver biopsy specimen showed that some hepatocytes were positive for 4-hydroxy-2-nonenal (HNE) (Fig. 3a, d) and C-EBP homologous protein (CHOP) (Fig. 3b, e). The expression of p62 increased in some hepatocytes (Fig. 3c). Staining for p62 also revealed Mallory-Denk bodies (MDBs) (Fig. 3f, arrows). To confirm genetic diagnosis, informed consent according to the Ethics Committee of the University of Occupational and Environmental Health (confirmation No.: H28-03) was obtained before taking blood sample for genes analysis. Gene analysis revealed a heterozygous mutation in the SLC40A1 gene [a heterozygous A > C transition at c.470 in exon 5 (p.D157A) resulting in a change in the amino acid at codon 157 from Asp to Ala] (Fig. 4a). Moreover, in the SLC40A1 gene, a heterozygous mutation at c.663 in exon 6 (p.V221V), which did not result in any amino acid substitution, was observed (Fig. 4b). No sequence alterations were found in the other genes associated with hemochromatosis including HFE, HJV, HAMP and TfR2. Thus, we diagnosed the patient as type 4 hereditary hemochromatosis due to heterozygous SLC40A1 mutation. We analyzed his daughter’s gene after obtained the informed consent, but her SLC40A1 was normal. Because he was intolerance to phlebotomy therapy due to anemia, we started deferasirox (12 mg/kg/day) treatment. After starting this chelating therapy, elevation of serum ferritin level and aminotransferase levels gradually improved (Fig. 4c).

Computed tomography (CT) and magnetic resonance imaging (MRI) image of this patient. Head and abdominal CT showed increase in density of pituitary gland (arrowhead) (a, b), liver and spleen (c). Head and abdominal MRI showed a significant decrease in intensity of the pituitary gland (arrowhead) (d, e), liver and spleen (f) on the T2-weighted images

Histopathological findings of the liver biopsy specimen. a, b Hematoxylin and eosin staining showed moderate inflammatory infiltrate and brown pigmentation in hepatocytes. c Masson trichrome staining showed severe fibrosis. d Berlin blue staining showed iron accumulation, located both in hepatocytes and sinusoidal lining cells. Inset shows higher magnification to discriminate sinusoidal lining cells (arrowheads) from hepatocytes. The hepatocytes with diffuse iron deposits were seen. Scale bars: 200 µm (a, c, d: ×100 magnification), 100 µm (b: ×200 magnification), and 50 µm (d: inset)

Immunohistochemistry of the liver biopsy specimen for 4-hydroxy-2-nonenal (HNE) (a, d), C-EBP homologous protein (CHOP) (b, e), and p62 (c, f). a, b Some hepatocytes were positive for oxidative stress marker (HNE) and ER stress marker (CHOP). c The expression of p62 increased in some hepatocytes. f Mallory-Denk bodies were detected clearly in p62 staining (arrows). d, e The expression of CHOP and HNE in the cells with Mallory-Denk body decreased (arrows). Scale bars: 200 μm (a, b, c: × 200 magnification) and 100 μm (d, e, f: × 640 magnification)

The sequencing electrogram and the clinical course of this patient. a Identification of the p.D157A mutation. Electropherogram showing the presence of heterozygous c.470 A > C on exon 5 of the SLC40A1. b Identification of the p.V221V mutation. Electropherogram showing the presence of heterozygous c.663 T > C on exon 6 of the SLC40A1. The arrows indicate the site of mutation. c Deferasirox treatment successfully decreased serum ferritin level as well as serum aspartate transaminase (AST) and alanine aminotransferase (ALT) levels
Discussion
Identification of genes implicated in iron transport and storage has induced reclassification and genetic distinction of hemochromatosis [12]. SLC40A1 gene mutation-caused type 4 hemochromatosis falls into two functional categories, underlying two distinct clinical entities (type 4A hemochromatosis or classical ferroportin disease and type 4B hemochromatosis or ferroportin-associated hemochromatosis) [13]. The majority of pathogenic variants, defined as “loss of function” mutations since they lead to decreased iron export from cells, give rise to type 4A hemochromatosis, an atypical form of primary iron overload, characterized by iron overload localized mainly in Kupffer cells and macrophages [14, 15]. The phenotypic features of type 4A hemochromatosis demonstrate that early elevation of serum ferritin, normal or mild elevation of serum iron, low to normal transferrin saturation, iron deposition in the Kupffer cells, iron overload in the spleen (black spleen in T2-weighted MRI), and aggressive phlebotomy regimens can be a problem due to borderline anemia [16]. On the other hand, “gain-of-function” mutations result in higher serum iron concentrations due to abnormal iron release from iron-recycling macrophages and enterocytes which manifest in elevated transferrin saturation. So phenotypic features of type 4B hemochromatosis are marked elevation of transferrin saturation, iron deposition in hepatocytes and commonly lack of iron in Kupffer cells, diffuse iron deposition in the liver but not spleen in MRI, and tolerance to phlebotomy [17].
Various SLC40A1 mutations had been identified and involved in iron binding, intracellular gate interaction and iron egress, respectively [18, 19]. Thus, abnormalities of ferroportin caused by SLC40A1 mutations have been considered to cause wide clinicopathological heterogeneity, showing marked differences in type of target organ and iron deposition, wide spectrum of sub-phenotypes and degree of penetrance [20,21,22].
In the present case, serum ferritin and transferrin saturation significantly elevated. Head CT and MRI showed enlargement and iron overload in the pituitary gland (Fig. 1). Endocrine examination revealed hypothyroidism, hypogonadism and growth hormone deficiency in addition to adrenal insufficiency (Table 1). Because pituitary functional challenge test demonstrated no or low response, we diagnosed him as anterior pituitary dysfunction (Table 2). Abdominal CT and MRI showed iron overload in the liver and spleen (Fig. 1). Liver biopsy specimen revealed marked iron deposition in hepatocytes as well as sinusoidal lining cells (probably Kupffer cells) (Fig. 2). Previous reports have demonstrated that liver biopsy shows iron overload predominates in Kupffer cells in type 4A, whereas in hepatocytes in type 4B hemochromatosis [21]. Thus, we focused on iron deposition in hepatocytes and Kupffer cells. Thus, hypopituitarism and liver dysfunction of this patient were considered to be caused by severe iron deposition in the pituitary gland and liver. Immunohistochemical analysis revealed increase of p62 expression and formation of MDB (Fig. 3c, f). Most p62-positive cells were positive for HNE, an oxidative stress marker, and CHOP, an ER stress marker (Fig. 3a, b, c). When contrast to the findings of Berlin blue staining (Fig. 2d), the hyper positive area of HNE and CHOP was similar in iron deposition area (Fig. 3a, b). Furthermore, the expression of HNE and CHOP seemed to be weaker in the cells with MDB than that of the cells without MDB (Fig. 3d, e, f, arrows). MDBs are hepatocellular inclusions observed in several liver disorders and seen as irregularly shaped cytoplasmic inclusions typically located in proximity to nucleus. Keratin 8 and 18, ubiquitin, chaperones and p62 are the major components of MDBs. MDB formation has been considered to result from an excess burden of misfolded protein accumulation and sequestration of these abnormal proteins in the cytoplasm of hepatocytes [23]. We previously reported that MDB formation could protect hepatocytes from ER stress and oxidative stress by sequestering abnormal proteins [24, 25]. Thus, MDB formation may improve the hyper iron deposition-induced oxidative stress in the patients with hemochromatosis. After treatment of deferasirox, the level of aminotransferase and ferritin gradually decreased (Fig. 4c). Although hepatic iron overload is well known to induce oxidative stress by generating reactive oxygen species (ROS) [26], the effect of iron overload on ER stress has not been fully understood. Our results demonstrated that excess iron deposition induced ER stress in hepatocytes. Thus, anti-ER stress agents, such as chemical chaperones, may be useful for treatment of the patients with hemochromatosis who are intolerance to phlebotomy or iron chelating therapy. Iron overload in pituitary gland of this patient is seemed to be secondary to serum iron burden and is mostly associated with pituitary dysfunction. So “gain of function” type seemed to be applied to the present case by the clinical and pathological findings.
To date, SLC40A1-linked hemochromatosis had been found only in four patients of three families in Japan, including both type 4A and type 4B hemochromatosis [27, 28], except the present case. Among them, two patients, a family with type 4A hemochromatosis, had c.1467 A > C heterozygous mutation (p.R489S) [29]. The other two patients were presented as type 4B hemochromatosis. One of them had n.117A > G heterozygous mutation [30] and c.470 A > C heterozygous mutation (p.D157A) [28], the same mutation in the present case. So, this case was the fifth patient with type 4 hemochromatosis in Japan. Prior study demonstrated that the similar mutation in ferroportin (p.D157G) showed significantly reduced expression in cultured human embryonic kidney 293 T (HEK293T) cells, while the localization was normally to the cell surface [15]. The Caucasian patient with the same heterozygous mutation in the SLC40A1 gene (p.D157A) had been reported to demonstrate the phenotype of “loss of function”, type 4A hemochromatosis [16, 31]. On the other hand, the heterozygous mutation in the SLC40A1 gene (p.D157A) in two Japanese patients, including this patient, caused the phenotype of “gain of function”, type 4B hemochromatosis. Thus, detailed functional assay seems to be needed for elucidating whether this mutation (p.D157A) in the SLC40A1 gene induces “loss of function” or “gain of function”. Previous report demonstrated that some of the same mutation described with both phenotypes of type 4 hemochromatosis due to environmental co-factors [32]. Interestingly, this case presented anterior pituitary function deficiency, but not the other organ symptoms, including as glucose intolerance, heart failure and skin pigmentation. Another Japanese patient with type 4B hemochromatosis was reported to present glucose intolerance and skin pigmentation, but not pituitary dysfunction [28]. Therefore, further examination should be needed to investigate the symptomatic heterogeneity in this SLC40A1 gene mutation.
Conclusion
We described a case of type 4B hemochromatosis with heterozygous mutation in the SLC40A1 gene (p.D157A). Type 4 hereditary hemochromatosis could cause wide clinicopathological heterogeneity. We should suspect the possibility of hereditary hemochromatosis even in Japanese patients with severe iron overload. And a mutation of SLC40A1 gene (p.D157A) causes gain of function of ferroportin.
Abbreviations
- ACTH:
-
Adrenocorticotropic hormone
- ADH:
-
Antidiuretic hormone
- ALT:
-
Alanine aminotransferase
- ALP:
-
Alkaline phosphatase
- AMA:
-
Anti-mitochondrial antibody
- AST:
-
Aspartate transaminase
- CHOP:
-
C-EBP homologous protein
- CT:
-
Computed tomography
- CRH:
-
Corticotropin-releasing hormone
- ECG:
-
Electrocardiogram
- FSH:
-
Follicle-stimulating hormone
- FPG:
-
Fasting plasma glucose
- γ-GTP:
-
γ-Glutamyltransferase
- GH:
-
Growth hormone
- GHRP2:
-
Growth hormone-releasing peptide 2
- HEK293T:
-
Human embryonic kidney 293T
- HNE:
-
4-Hydroxy-2-nonenal
- HOMA-IR:
-
Homeostasis model assessment of insulin resistance
- IGF-1:
-
Insulin-like growth factor-1
- IQR:
-
Interquartile range
- LH:
-
Luteinizing hormone
- LHRH:
-
Luteinizing hormone-releasing hormone
- M2BPGi:
-
Mac-2 binding protein glycosylation isomer
- MDB:
-
Mallory-Denk body
- MRI:
-
Magnetic resonance imaging
- PRL:
-
Prolactin
- ROS:
-
Reactive oxygen species
- TIBC:
-
Total iron binding capacity
- TSH:
-
Thyroid-stimulating hormone
- UIBC:
-
Unsaturated iron binding capacity
References
Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS (2011) Diagnosis and Management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54:328–343
Powell LW, Seckington RC, Deugnier Y (2016) Haemochromatosis. Lancet 388:706–716
European Association For The Study Of The Liver (2010) EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol 53:3–22
McDonald CJ, Wallace DF, Crawford DH, Subramaniam VN (2013) Iron storage disease in Asia-Pacific populations: the importance of non-HFE mutations. J Gastroenterol Hepatol 28:1087–1094
Njajou OT, Vaessen N, Joosse M, Berghuis B, van Dongen JW, Breuning MH, Snijders PJ, Rutten WP, Sandkuijl LA, Oostra BA, van Duijn CM, Heutink P (2001) A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet 28:213–214
Speletas M, Kioumi A, Loules G, Hytiroglou P, Tsitouridis J, Christakis J, Germenis AE (2008) Analysis of SLC40A1 gene at the mRNA level reveals rapidly the causative mutations in patients with hereditary hemochromatosis type IV. Blood Cells Mol Dis 40:353–359
Zhang DL, Wu J, Shah BN, Greutélaers KC, Ghosh MC, Ollivierre H, Su XZ, Thuma PE, Bedu-Addo G, Mockenhaupt FP, Gordeuk VR, Rouault TA (2018) Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk. Science 359:1520–1523
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306:2090–2093
Pietrangelo A (2010) Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology 139:393–408
Drakesmith H, Nemeth E, Ganz T (2015) Ironing out Ferroportin. Cell Metab 22:777–787
Ka C, Guellec J, Pepermans X, Kannengiesser C, Ged C, Wuyts W, Cassiman D, de Ledinghen V, Varet B, de Kerguenec C, Oudin C, Gourlaouen I, Lefebvre T, Férec C, Callebaut I, Le Gac G (2018) The SLC40A1 R178Q mutation is a recurrent cause of hemochromatosis and is associated with a novel pathogenic mechanism. Haematologica 103:1796–1805
Griffith WJ (2007) Review article: the genetic basis of hemochromatosis. Aliment Pharmacol Ther 26:331–342
Brissot P, Loreal O (2016) Iron metabolism and related genetic diseases: a cleared land, keeping mysteries. J Hepatol 64:505–515
Pietrangelo A (2004) The ferroportin disease. Blood Cells Mol Dis 32:131–138
De Domenico I, Ward DM, Nemeth E, Vaughn MB, Musci G, Ganz T, Kaplan J (2005) The molecular basis of ferroportin-linked hemochromatosis. Proc Natl Acad Sci USA 102:8955–8960
Pietrangelo A (2017) Ferroportin disease: pathogenesis, diagnosis and treatment. Haematologica 102:1972–1984
Mayr R, Janecke AR, Schranz M, Griffiths WJH, Vogel W, Pietrangelo A, Zoller H (2010) Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol 53:941–949
Callebaut I, Joubrel R, Pissard S, Kannengiesser C, Gérolami V, Ged C, Cadet E, Cartault F, Ka C, Gourlaouen I, Gourhant L, Oudin C, Goossens M, Grandchamp B, De Verneuil H, Rochette J, Férec C, Le Gac G (2014) Comprehensive functional annotation of 18 missense mutations found in suspected hemochromatosis type 4 patients. Hum Mol Genet 23:4479–4490
Vlasveld LT, Janssen R, Bardou-Jacquet E, Venselaar H, Hamdi-Roze H, Drakesmith H, Swinkels DW (2019) Twenty years of ferroportin disease: a review or an update of published clinical, biochemical, molecular, and functional features. Pharmaceuticals (Basel) 12:E132
Mayr R, Griffiths WJ, Hermann M, McFarlane I, Halsall DJ, Finkenstedt A, Douds A, Davies SE, Janecke AR, Vogel W, Cox TM, Zoller H (2011) Identification of mutations in SLC40A1 that affect ferroportin function and phenotype of human ferroportin iron overload. Gastroenterology 140:2056–2063
Zhang W, Lv T, Huang J, Ou X (2017) Type 4B hereditary hemochromatosis associated with a novel mutation in the SLC40A1 gene: a case report and a review of the literature. Medicine (Baltimore) 96:e8064
Majore S, Bonaccorsi di Patti MC, Valiante M, Polticelli F, Cortese A, Di Bartolomeo S, De Bernardo C, De Muro M, Faienza F, Radio FC, Grammatico P, Musci G (2018) Characterization of three novel pathogenic SLC40A1 mutations and genotype/phenotype correlation in 7 Italian families with type 4 hereditary hemochromatosis. Biochim Biophys Acta Mol Basis Dis 1864:464–470
Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM, Cardin M, Omary MB (2007) From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res 313:2033–2049
Honma Y, Harada M, Sato M, Katsuki Y, Hiura M, Shibata M, Narita R, Harada R, Abe S, Tabaru A, Tajiri N, Shimajiri S (2011) Late diagnosed Wilson disease with hepatic and neurological manifestations. Hepatol Res 41:270–276
Harada M, Hanada S, Toivola DM, Ghori N, Omary MB (2008) Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology 47:2026–2035
Nishina S, Hino K, Korenaga M, Vecchi C, Pietrangelo A, Mizukami Y, Furutani T, Sakai A, Okuda M, Hidaka I, Okita K, Sakaida I (2008) Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology 134:226–238
Hayashi H, Wakusawa S, Motonishi S, Miyamoto K, Okada H, Inagaki Y, Ikeda T (2006) Genetic background of primary iron overload syndrome in Japan. Intern Med 45:1107–1111
Yamashita T, Morotomi N, Sohda T, Hayashi H, Yoshida N, Ochi K, Ohkura I, Karita M, Fujiwara H, Yamashita H, Hattori A, Tatsumi Y (2014) A male patient with ferroportin disease B and a female patient with iron overload similar to ferroportin disease B. Clin J Gastroenterol 7:260–264
Koyama C, Wakusawa S, Hayashi H, Ueno T, Suzuki R, Yano M, Saito H, Okazaki T (2005) A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern Med 44:990–993
Liu W, Shimomura S, Imanishi H, Iwamoto Y, Ikeda N, Saito M, Ohno M, Hara N, Yamamoto T, Nakamura H, Hada T (2005) Hemochromatosis with mutation of the ferroportin 1 (IREG1) gene. Intern Med 44:285–289
Saja K, Bignell P, Robson K, Provan D (2010) A novel missense mutation c.470 A>C (p. D157A) in the SLC40A1 gene as a cause of ferroportin disease in a family with hyperferritinemia. Br J Haematol 149:914–916
Le Lan C, Mosser A, Ropert M, Detivaud L, Loustaud-Ratti V, Vital-Durand D, Roget L, Bardou-Jacquet E, Turlin B, David V, Loréal O, Deugnier Y, Brissot P, Jouanolle AM (2011) Sex and acquired cofactors determine phenotypes of ferroportin disease. Gastroenterology 140:1199–1207
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Honma, Y., Karasuyama, T., Kumamoto, K. et al. Type 4B hereditary hemochromatosis due to heterozygous p.D157A mutation in SLC40A1 complicated with hypopituitarism. Med Mol Morphol 54, 60–67 (2021). https://doi.org/10.1007/s00795-020-00259-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00795-020-00259-1