
Abstract
Demyelination leads to axonal changes that involve the functions and dynamics of axonal mitochondria supporting metabolism and survival of axons. However, the changes in the physical interactions between mitochondria and endoplasmic reticulum, called mitochondria-associated membranes, are poorly understood in demyelinated axons. In this study, we investigated the three-dimensional ultrastructural changes in membrane juxtapositions between mitochondria and endoplasmic reticulum in axons of a chronic progressive demyelination mouse model caused by extra copies of proteolipid protein (PLP4e). In the optic nerve of PLP4e mice, most axons were ensheathed by myelin by age 1 month, but were demyelinated by age 5 months. At age 1 month, mitochondria in PLP4e mice were slightly larger than those in wild-type mice, while the size and frequency of juxtaposition were similar. At age 5 months, the sizes of mitochondria and size of juxtaposition in PLP4e mice were prominently larger than those in wild-type mice. In degenerating axons under demyelination, the enlargement of mitochondria was diminished, while the density and frequency of juxtaposition were similar to those of non-degenerating axons. These results suggest that interactions between mitochondria and ER are enhanced in chronically demyelinated axons and maintained during axonal degeneration in hereditary myelin diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Demyelination leads to structural and molecular changes in axons, and has been implicated in the axonal degeneration that contributes to the permanent neurological deficits seen in demyelination disorders [1, 2]. Under demyelination, axolemmal proteins such as ion channels are redistributed and axons are exposed to the microenvironment, which is often inflammatory and can damage axonal molecules [3, 4]. Metabolic and environmental changes of axons are associated with functional modulations of axonal organelles including mitochondria, which involve alterations in their transport, localization and morphology [5,6,7,8]. These behavioral aspects of mitochondria are controlled by molecules associated with cytoskeletons or inner and outer mitochondrial membranes [9]. Since mitochondrial function is essential for axonal integrity and survival, the interactions between mitochondria and other organelles, which are critical for the functional maintenance of mitochondria and axons, may play a role in the pathophysiology of demyelinating disorders.
The interactions between mitochondria and endoplasmic reticulum (ER), which are referred to as the mitochondria-associated membrane (MAM), play several roles in cellular metabolism and organelle dynamics, such as lipid metabolism, Ca2+ signaling and mitochondrial fission [10,11,12,13,14]. These interactions are characterized by membrane juxtaposition with a distance of 10–30 nm, and are associated with complexes that include molecules on mitochondrial and/or ER membranes, such as Mfn2 [15,16,17]. Impaired interactions between mitochondria and ER have been implicated in the degeneration of neurons and axons in neurological disorders, and are associated with mitochondrial dysfunctions in a myelin disease model [18,19,20]. However, the structural changes and roles of mitochondria–ER interactions in demyelinated axons are still poorly understood.
In this study, three-dimensional ultrastructural changes in the membrane juxtaposition between mitochondria and ER in demyelinated axons were investigated using serial block-face scanning electron microscopy (SBF-SEM), where serial electron microscopic images were acquired from optic nerve tissues and all of the membrane juxtapositions of individual mitochondria and ER in axons were reconstructed. The structural parameters of membrane juxtapositions such as number, area, and relationship to the mitochondrial surface area were analyzed in a mouse model of chronic demyelination, which leads to almost complete and reproducible demyelination in optic nerves following myelin formation. The results demonstrated that enlargement of the membrane juxtaposition of mitochondria and ER is caused by the chronic loss of myelin. Furthermore, the results suggested that the interaction of mitochondria and ER could be maintained in axonal degeneration in hereditary demyelinating disorders.
Materials and methods
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Yamanashi and National Institute for Physiological Sciences and performed in accordance with institutional guidelines. Mice with extra copies of the PLP gene, PLP4e/− mice (PLP4e) were obtained from Riken BRC (Tsukuba, Ibaraki, Japan) and maintained by crossing with DBF-1 mice purchased from Japan SLC (Shizuoka, Japan) [21]. Male PLP4e mice and control littermates were used in all experiments.
Observations and image analyses with SBF-SEM
16 mice were anaesthetized by pentobarbital and transcardially perfused with 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1M phosphate buffer (pH 7.4). Optic nerves were collected and immersed in the same fixatives overnight, and samples in resin for SBF-SEM observation were prepared as previously described [7, 22]. Serial images were acquired with a field emission scanning electron microscope (Merlin or Sigma from Carl Zeiss, Germany) equipped with 3View, a system with a built-in ultramicrotome and a back-scattered electron detector (Gatan, CA, USA), at a resolution of approximately 6 nm/pixel in the X and Y directions and 50–70 nm in the Z-direction. The 500–720 serial images from each dataset were handled with ImageJ with Fiji plugins (https://fiji.sc/), and manual segmentation, reconstruction and analyses were performed with TrakEM2 [23] and Amira (FEI Visualization Science Group, Hillsboro, OR, USA).
In the serial electron microscopic images, mitochondria were identified as cylindrical double-membrane structures with interior crista membranes [24]. ER was identified as tubular and continuous cisternae forming networks throughout the cytoplasm [20]. Axons were identified by the presence of endoplasmic reticulum enriched beneath the axolemma, and abundant and evenly distributed cytoskeletal profiles in the axoplasm [25]. The membrane juxtapositions between mitochondria and ER were defined as the close membrane apposition of mitochondria and ER where there is not more than 15 nm of space between the two membranes on electron micrographs. Degenerating axons were identified as beading axonal profiles with occasional axonal swellings and swollen mitochondria with a large matrix and sparse interior cristae.
Triple immunostaining and measurement of fluorescence intensity
Optic nerves were obtained from three wild-type and 3 PLP4e mice following transcardial perfusion with and subsequent overnight incubation in 0.1M phosphate buffer containing 4% paraformaldehyde. The tissues were immersed in phosphate buffered saline (PBS) containing 15% sucrose at 4 °C for 4 h, and then in PBS containing 30% sucrose and 5% glycerol at 4 °C for 2 days. Then tissues were frozen in OCT compound, sectioned at the thickness of 5–14 µm using cryostat (CM3050 Leica, Wetzlar, Germany), and attached onto Matsunami adhesive silane (MAS)-coated glass slides (Matsunami glass Ind., Ltd., Osaka, Japan, S9226). The sections were washed in PBS, and microwaved in 0.01M citrate buffer three times without boiling for antigen retrieval. After cooling down for 20 min at room temperature, sections were incubated in PBS containing 0.5% Triton-X 100 at room temperature for 2 h, and then in PBS containing 2% bovine serum albumin (BSA, Sigma-Aldrich Japan, Tokyo, Japan) and the mixture of primary antibodies at 4 °C overnight. The primary antibodies were against neurofilament (Novus Biologicals, Littleton, USA, NB300-217, 1:4000), Tom20 (Santa Cruz Biotechnologies, Santa Cruz, USA, sc-11415, 1:1000), and mitofusin 2 (Mfn2, Abcam, Cambridge, United Kindom, ab56889, 1:1000). Then sections were washed in PBS, and incubated in PBS containing 2% BSA and fluorescent secondary antibodies (Cy5-conjugated goat anti-mouse IgG 1:600 (Chemicon, Temecula, USA, AP124S), Alexa488-conjugated goat anti-rabbit IgG 1:800 (Thermo Fisher Scientific, Rockford, USA, A11034), Alexa594-conjugated goat anti-chicken IgG 1:800 (Thermo Fisher Scientific, A11042)) at room temperature for 3 h in dark. After washing and embedding, the immunostained sections were observed using an A1R confocal microscope with a 100× objective lens (Nikon Corporation, Tokyo, Japan).
The fluorescence intensity of immunostaining for TOM20 and Mfn2 was measured in 16bit images using ImageJ with Fiji plugins (http://fiji.sc/wiki/index.php/Fiji). The mean fluorescence intensity (mFa) was measured in each neurofilament-positive axonal profile, and mean fluorescence intensity of the background (mFb) was measured in the adjacent regions negative for immunostaining. The fluorescence intensity was calculated with the following formula: mFa − mFb.
Western blotting
Optic nerve tissues were homogenized and lysed in buffer containing 40 mM Tris–HCl, 5 mM EGTA, 100 mM NaCl, 1% Triton-X 100 and protease inhibitors which included phenylmethylsulfonyl fluoride (50 µg/ml), pepstatin A (10 µg/ml), leupeptin (10 µg/ml), ethylenediaminetetraacetic acid (1–5 mM), N-ethylmaleimide (1 mM). Protein concentrations were determined by Bradford protein assay (Bio-Rad Laboratories, Hercules, USA). Proteins were loaded, electrophoresed on 4–15% acrylamide gel and transferred onto polyvinylidene fluoride (PVDF) membrane which has a nominal pore size 0.45 µm. The bands were visualized with anti-Mfn2 (Abcam, ab56889, 1:5000) and anti-voltage dependent anion channel 1 (VDAC1, Abcam, ab 14734, 1:5000) primary antibodies, the horse radish peroxidase-conjugated goat anti-mouse IgG secondary antibody, and ECL-prime Western blot detection reagent (GE healthcare Life Sciences, Uppsala, Sweden).
Quantification of western blots for Mfn2 and VDAC1 was done by measuring band intensities with subtraction of the background, and dividing the intensity of Mfn2 with the intensity of VDAC1 in each blot, using ImageJ.
Statistical analysis
SBF-SEM analyses and measurements were performed using 3 WT and PLP4e mice at age 1 month and 5 months, and 2 WT and PLP4e mice at age 3 months. Statistical comparisons and normality tests were performed using Prism (GraphPad Software, La Jolla, CA, USA). Graphs show medians (bars), quartile ranges (boxes), and 10–90% ranges (whiskers), and statistical comparisons were performed using the MannWhitney U test or t test. Multiple comparisons were made with Bonferroni corrections.
Results
To examine mitochondrial changes and their interactions with ER in chronically demyelinated axons, we used mice with extra copies of the PLP gene, PLP4e/− mice (PLP4e), which exhibit chronically progressive demyelination in the central nervous system [21]. To confirm chronic demyelination in the optic nerves of PLP4e mice, we performed ultrastructural analyses of optic nerves of wild-type mice (WT) and PLP4e, using SBF-SEM. Axons in the optic nerves of WT were largely myelinated at age 1 month (mo) (Fig. 1a), and 3 mo (Fig. 1b), and almost fully myelinated at 5 mo (Figs. 1c, 2a). In contrast, although comparable numbers of optic nerve axons were myelinated in PLP4e at 1 mo (Fig. 1d), the number of myelinated axons was decreased at 3 mo (Figs. 1e, 2a). In 5 mo-PLP4e mice, almost all axons were demyelinated and lacked a myelin sheath (Figs. 1f, 2a). In PLP4e mice, the G-ratio (ratio of axonal diameter-to-the outer diameter of the myelin sheath), which is higher when the myelin sheath is thinner, was higher than that in WT mice at 1, 3, and 5 mo (Fig. 2b–e). These results reflect myelin formation followed by progressive and almost complete demyelination in optic nerves of PLP4e mice.

Chronically progressive demyelination in PLP4e optic nerves. Most axons (Ax) were myelinated (arrowheads) in wild-type (WT) mice at 1 (a, 1 mo), 3 (b, 3 mo) and 5 months of age (c, 5 mo). Many axons were myelinated in PLP4e mice at 1 mo (d, arrowheads). In PLP4e, some axons were demyelinated at 3 mo (e, arrows), while others remained myelinated (e, arrowheads). At 5 mo, most axons were demyelinated (f, arrows). Bars: 1 µm

Axonal ensheathment by thinner myelin and prominent demyelination in PLP4e optic nerves. The percentage of myelinated axons (a) and G-ratio (ratio of the axonal diameter-to-the outer diameter of the myelin sheath), b in wild-type (WT) and PLP4e mice at age 1, 3, and 5 months (mo) are shown. The relationship between axon diameter and G-ratio (c–e) is also compared at 1 (c), 3 (d), and 5 mo (e). *p < 0.05, ***p < 0.001 by the Mann–Whitney U test. N = 12 images (a), 150 axons (b), 1 and 5 mo (c, e) or 100 axons (b, 3 mo, d)
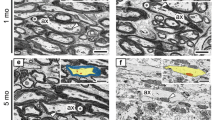
We next examined how mitochondrial morphology and distribution are altered during the chronically progressive demyelination of PLP4e. Axons and their mitochondria in optic nerves of WT and PLP4e mice at 1, 3, and 5 mo were clearly identified in single SBF-SEM images, and their 3D reconstruction showed the sizes and distribution of all mitochondria in individual axons (Figs. 3, 4, 5). In PLP4e mice at 5 mo, while some axons were not degenerative and included large mitochondria with dense cristae (Fig. 4d–f, l), some axons exhibited beading and contained swollen and round mitochondria, and these axons were separately analyzed as degenerating axons (Fig. 4g–j). At 1 mo, the % axonal volume occupied by mitochondria (mtVol/axVol) and number of mitochondria per unit axonal volume (mtNo/axVol) were not significantly different between WT and PLP4e, while the volume of individual mitochondria (mtVol) was slightly larger in PLP4e mice (Figs. 3a–f, 5a–c). In contrast, mtVol/axVol was significantly higher in axons of PLP4e optic nerve at 3 mo, and this increase was attributable to the increased mtNo/axVol ratio (Figs. 3g–l, 5a–c). In addition, mtVol/axVol in demyelinated axons of PLP4e mice at 5 mo was significantly higher than those of PLP4e mice at 3 mo and WT mice at 5 mo (Figs. 4a–f, k, l, 5a). The higher mtVol/axVol ratio in demyelinated axons of PLP4e mice at 5 mo was due to the prominent increase in mtVol, since, at 5 mo, mtNo/axVol in WT was similar to that in PLP4e mice (Fig. 5b, c). The increase of mitochondrial volume in non-degenerating axons with demyelination in 5 mo PLP4e mice was not due to mitochondrial swelling, since neither enlargement of the mitochondrial matrix nor sparse cristae was observed in mitochondria of non-degenerating axons in PLP4e mice (Fig. 4l), as in mitochondria of myelinated axons in WT mice (Fig. 4k). In PLP4e mice at 5 mo, mtVol in degenerating axons were lower than those in non-degenerating axons, while mtVol/axVol were similar (Fig. 5a–c). The lower mtVol in degenerating axons is likely to be due to the enhanced mitochondrial fission along with mitochondrial swelling, since mitochondria are cylindrical with compact matrix in non-degenerating axons but appear smaller and more spherical with sparse cristae and enlarged matrix in degenerating axons (Fig. 4f, g, j). Collectively, these results suggest that axonal volume occupied by mitochondria is dramatically increased under chronically progressive demyelination, and involves mitochondrial fusion leading to the enlargement of individual mitochondria. Furthermore, the degeneration of chronically demyelinated axons is associated with a decrease in the axonal volume occupied by mitochondria and the sizes of individual mitochondria.

Three-dimensional reconstruction of axonal mitochondria in optic nerve axons of wild-type (WT) and PLP4e mice at age 1 and 3 months. Original electron microscopic images (a, d, g, j), labeled images (b, e, h, k) and reconstructions (c, f, i, l) of axons (gray) and mitochondria (various colors) in WT (a–c, g–i) and PLP4e (d–f, j–l) mice at age 1 (a–f) and 3 (g–l) months are shown. Bars: 1 µm

Three-dimensional reconstruction of axonal mitochondria in optic nerve axons of wild-type (WT) and PLP4e mice at age 5 months. Original electron microscopic images (a, d, g, h), labeled images (b, e, i) and reconstructions (c, f, j) of axons (gray) and mitochondria (various colors) in WT (a–c) and PLP4e (d–j) mice at age 5 months are shown. In PLP4e mice, an axon (Ax) with degenerative changes, including axonal swelling and fragmented and swollen short mitochondria (g, arrowheads), and high magnification of the swollen mitochondrion with sparse cristae (g, inset) are also shown. A mitochondrion in a myelinated axon of a WT mouse (k) and a large mitochondrion in non-degenerating demyelinated axon of a PLP4e mouse (l) at age 5 months are shown. Bars: 1 µm (a–j) and 0.5 µm (g, inset, k–l)

Increased mitochondrial volume in chronically demyelinated axons and decreased mitochondrial sizes in degenerative axons in optic nerves of PLP4e mice. Graphs of mitochondrial volume per axonal volume (mtV/axV, a), mitochondrial number per axonal volume (mtNo/axV, b), and sizes of individual mitochondria (c) are shown. Data in PLP4e mice at age of 5 months are obtained from non-degenerating (nm) and degenerating (de) axons. N (WT-1mo, PLP4e-1mo, WT-3mo, PLP4e-3mo, WT-5mo, PLP4e-5mo, degenerating axons in PLP4e-5mo) = 9, 9, 6, 6, 9, 9, 9 axons (a, b) or 102, 94, 112, 114, 174, 217 and 183 mitochondria (c) *p < 0.05, **p < 0.01, ***p < 0.001, n.s. not significant by the Mann–Whitney U test
Maintenance of mitochondrial functions requires physical interactions with ER through MAM. To examine whether the physical interactions between mitochondria and ER are modulated due to the changes in mitochondria under chronic demyelination, the membrane juxtapositions of mitochondria and ER were reconstructed and analyzed in the myelinated and demyelinated axons of WT and PLP4e mice, using SBF-SEM. The physical contacts (< 15 nm) between mitochondria and ER were identified (Fig. 6a, b, d, e, g, h) and segmented as MAM in serial electron microscopic images obtained by SBF-SEM (Fig. 6c, f, i). The contact areas were measured along with structural parameters of mitochondria and ER in the reconstructed 3D volume of axoplasm (Fig. 6j–m). The surface areas of individual mitochondria (miA) in myelinated axons of PLP4e mice were slightly larger than those of WT mice at 1 mo (Fig. 7a). However, the total MAM area for individual mitochondria (Fig. 7b) was similar in myelinated axons of WT and PLP4e at 1 mo. In contrast, at 5 mo, miA as well as the total areas of MAM for individual mitochondria in demyelinated axons of PLP4e mice were significantly larger than those in myelinated axons of WT mice (Fig. 7a, b). The larger MAM areas in demyelinated axons of PLP4e mice were not simply due to the enlargement of mitochondria (increase of miA), since the MAM area per unit mitochondrial surface area (% MAM area) in PLP4e was also larger than that in WT (Fig. 7d). The larger % MAM area in PLP4e was considered to be attributable to the enlargement of each MAM (Fig. 7f). This enlargement of MAM is not related to the total length of ER in axoplasm, since the measured ER length per unit axoplasmic volume (ER length/axV) was similar in WT and PLP4e (Fig. 7g). In myelinated axons of 5 mo WT mice, the total MAM area appeared to be proportional to miA in each mitochondrion (Fig. 7h), while % MAM area was variable (Fig. 7i). In 5 mo PLP4e mice, a similar relationship was observed between miA and total MAM area (Fig. 7h), and % MAM area for some larger mitochondria appeared to be greater than that of similar-sized mitochondria in 5 mo WT (Fig. 7i). These results demonstrated that chronically progressive demyelination causes significant enhancement of mitochondria–ER interactions in optic nerve axons, and suggest that the enhancement under chronic demyelination is attributable to the enlargement of each MAM rather to an increased number of MAM.

Three-dimensional reconstructions of membrane juxtapositions between mitochondria and endoplasmic reticulum (ER) in optic nerve axons of wild-type (WT) and PLP4e mice at age 5 months. Serial electron microscopic images show mitochondria in an axon (Ax, a, d, g) and the juxtapositions of mitochondria and ER (b, c, e, f, h, i, arrowheads) of 166th (a–c), 167th (d–f) and 168th (g–i) images. The juxtapositions (c, f, i, red), mitochondria (c, f, i, yellow) and ER (c, f, i, blue) are colored. The areas marked with rectangles (a, d, g) are magnified (b, c, e, f, h, i). Three-dimensional reconstructions of mitochondria (j–m, yellow) and ER (j, l, light blue) and the juxtapositions between them (k, m, red, arrows) in WT (j, k) and PLP4e (l, m) mice at age 5 months are shown. Bars: 0.5 µm (a–i) and 1 µm (j–m)

The sizes of membrane juxtapositions between mitochondria and endoplasmic reticulum (ER), which are called mitochondria-associated membranes (MAM), are increased in demyelinated axons and maintained in degenerative axons in optic nerves of PLP4e. The graphs show the surface area of individual mitochondria (a), total area of MAM on individual mitochondria (b), number of MAM on individual mitochondria (c), % MAM area (d) and number (e) per unit mitochondrial surface area, size of each MAM (f) and total ER length per axonal volume (g) in wild-type (WT) and PLP4e mice at age 1 and 5 months (mo). For PLP4e mice at 5 mo, data from demyelinated axons with (de) or without (nm) degenerative changes are shown. The relationships between mitochondrial size and total MAM area (h) or MAM area per unit mitochondrial surface area (i) of individual mitochondria in WT and PLP4e mice at 5 mo are shown. Data in PLP4e mice at age of 5 months are obtained from non-degenerating (nm) and degenerating (de) axons. N (WT-1mo, PLP4e-1mo, WT-5mo, PLP4e-5mo, degenerating axons in PLP4e-5mo) = 102, 94, 174, 217 and 183 mitochondria (a–e), or 184, 240, 628, 1209 and 719 MAM (f). *p < 0.05, **p < 0.01, ***p < 0.001, n.s. not significant by the Mann–Whitney U test
In the demyelinated optic nerve tissues of PLP4e mice at 5 mo, a small number of axons were degenerating with an abnormal morphology including obvious beading and fragmented and swollen mitochondria (Fig. 4g–j). To examine whether there are changes in the interactions between mitochondria and ER in these degenerating axons, juxtapositions of the degenerating axons were reconstructed and compared with those of non-degenerating axons in the demyelinated optic nerves of PLP4e mice at 5 mo. Consistent with the mitochondrial fragmentation described above, mitochondria in the degenerating axons were smaller and had less miA compared with non-degenerating axons (Fig. 7a). In addition, degenerating axons had significantly smaller MAM on individual mitochondria than non-degenerating axons (Fig. 7b), although the % MAM area was similar in degenerating and non-degenerating axons (Fig. 7d). These results suggest that the degeneration of demyelinated axons is associated with mitochondrial fission and maintains juxtaposition of mitochondria and ER in optic nerves of PLP4e.
Regulation mechanisms of MAM areas under physiological as well as pathological circumstances are still not fully understood, but expression levels of molecules involved in MAM have been associated with the functional changes of MAM [17, 19, 26]. In particular, reduced expression of Mfn2 has been associated with increased interaction between ER and mitochondria [27, 28]. To address if expression level of Mfn2 is altered in demyelinated optic nerve axons of PLP4e, immunostaining for Mfn2, Tom20, a mitochondrial marker, and neurofilament, an axonal marker, was done in the optic nerve tissues obtained from 5 mo WT and PLP4e. The small dot-like immunoreactivities of Tom20 and Mfn2 were observed throughout the cross sections of optic nerves, and often co-localized with each other and that of neurofilament both in WT and PLP4e tissues (Fig. 8a, b). In the neurofilament-positive axonal profiles, the immunofluorescence intensities of Tom20 in WT and PLP4e mice were similar (Fig. 8c), while the immunofluorescence intensities of Mfn2 in PLP4e mice were higher than those in WT mice (Fig. 8d). To further examine the expression of Mfn2, we performed western blot of optic nerves of 5 mo WT and PLP4e mice. The results showed the expression of Mfn2 compared with a control mitochondrial protein, VDAC1, in PLP4e mice was higher than that in WT mice (Fig. 8e, f). These results suggest that the alteration of MAM under chronic demyelination of optic nerve axons in PLP4e accompanies increased expression level of axonal Mfn2.

Expression of Mfn2 is increased in demyelinated axons of PLP4e. Triple immunostaining for neurofilament (NF, blue), Tom20 (red) and mitofusin 2 (Mfn2, green) in optic nerves of wild-type (WT) and PLP4e mice at age of 5 months (mo) is shown (a, b). At the higher magnification (a, b, insets), areas demarcated with white lines indicated NF-immunopositive axons where Mfn2 and Tom20-positive mitochondria are co-localized in WT (a, arrowhead) and PLP4e (b, arrowheads) mice. The mean fluorescence intensities of Tom20 in WT and PLP4e mice are similar (c) but that of Mfn2 in PLP4e mice is significantly higher than that in WT mice (d). N = 420 NF-positive profiles. ***p < 0.001, ns not significant by the Mann–Whitney U test. Bars: 10\({\upmu }\text{m}\) and 1\({\upmu }\text{m}\) (insets). A.U. arbitrary unit. Western blotting analyses of Mfn2 and voltage-dependent anion channel 1 (VDAC1) in optic nerve tissues of WT and PLP4e mice (e) and their quantification (f) are shown. N = 3 mice for each genotype. **p < 0.01 by the t test
Discussion
This study examined how interactions between mitochondria and ER change in chronic demyelinated CNS axons in vivo, using SBF-SEM and a chronic demyelination model with extra copies of the Plp1 gene. The 3D reconstruction of serial electron microscopy images obtained from optic nerve tissues revealed that (1) MAM is enlarged in chronically demyelinated axons, (2) this increase is due to an increase in the size of individual MAM rather than in the density (number per mitochondrial area) of MAM, and (3) mitochondrial sizes are decreased but MAM sizes are similar in degenerating axons of demyelinated optic nerves. These results suggest that the sizes of MAM are actively regulated in response to the chronic loss of myelin, and are maintained upon axonal degeneration in demyelinating diseases.
In this study, 3D reconstruction of serial electron microscopic images was used to examine the 3D distribution of mitochondria–ER juxtaposition. Although light microscopic analysis of mitochondria and ER is a common approach for studying the spatial proximity of these organelles, light microscopy has limited resolution, and it can be difficult to determine if the distances between organelles are within the range of functional distance reported previously [16, 17, 29]. Electron microscopic analyses with 3D reconstruction can determine the distribution of all membrane juxtapositions in axonal segments, and how the number and sizes of MAM are independently regulated upon demyelination and axonal degeneration. Although electron microscopic analyses also have limitations, such as the impossibility of time-lapse observation, the results of this study support the notion that 3D ultrastructural analyses of mitochondria–ER interactions can provide unique information about their structural alterations in particular cellular compartments under physiological and pathological conditions.
In this study, the ultrastructural analysis of mitochondria–ER interactions was applied to a chronic and progressive demyelination mouse model with extra copies of the Plp1 gene. Pelizaeus–Merzbacher disease (PMD) and spastic paraplegia 2 (SPG2) in humans have been associated with mutations in PLP, and PMD is frequently caused by increased copies of PLP [30, 31]. The PLP4e mice in this study only mildly overexpress PLP, and therefore comparable myelination is followed by progressive and chronic demyelination [21, 32]. Although demyelination can be caused by various mechanisms, such as the inflammatory activation of immune cells and toxins that affect myelin-forming cells, changes in axonal organelles such as mitochondria are common in these disorders. Indeed, the mitochondrial volume/number is significantly increased upon demyelination regardless of its cause, and is likely to be related to the interaction with cytoskeletons [8, 33,34,35,36]. Our results are consistent with these studies, and therefore it is possible that enhanced interactions between mitochondria and ER could be commonly observed in demyelinated axons along with an increased volume of axonal mitochondria. Future studies will be needed to identify the changes in the interactions between mitochondria and ER upon demyelination caused by different mechanisms.
Changes in the interactions between mitochondria and ER have been associated with various mechanisms and molecules that are physically involved in this attachment or are associated with signaling that regulates these interactions. Mfn2 has been reported to be an important protein in the mitochondria–ER tether [37]. In mouse embryonic fibroblasts and HeLa cells, ablation of Mfn2 led to a decrease in ER–mitochondria interactions, while some studies have suggested that knockdown of Mfn2 increased ER–mitochondria contacts [15, 27, 28]. Mitochondrial ion channel, voltage-dependent anion channel, and inositol 1, 4, 5-trisphosphate receptor have been associated with the interactions between mitochondria and ER [38]. It is possible that multiple mechanisms regulate either the number or size of mitochondria–ER juxtapositions in axons. Our results suggest that the increased mitochondria–ER interactions upon demyelination can be modulated by an increase in the size of each interaction, but not by an increase in the number of interactions. Although we obtained data showing that increased axonal expression of Mfn2 is related to an increase in mitochondria–ER juxtaposition in demyelinated axons (Fig. 8), further studies using molecular approaches, such as knockdown of Mfn2 in vivo, will be needed to confirm that Mfn2 leads to altered ER–mitochondria connections in axons under demyelination.
Demyelination increased mitochondrial volume, which plays important roles in the survival of demyelinated axons [39]. The size of mitochondria–ER juxtaposition was proportional to the size of mitochondria in both myelinated axons and demyelinated axons, and the size of mitochondria–ER juxtaposition was significantly increased under chronic demyelination. These observations support the notion that enlarged mitochondria require larger mitochondria–ER interactions, and this enhanced mitochondria–ER interaction may help to protect against the degeneration and loss of axons. The regulation of mitochondria–ER interactions is important for mitochondrial functions, such as transfer of Ca2+ from ER to mitochondria, mitochondrial division and regulation of apoptosis [17]. Interactions between mitochondria and ER play important roles in the function and survival of neurons and axons. For example, impaired mitochondria–ER interactions have been reported in a mouse model with genetic mutations in myelin-forming genes, and are associated with mitochondrial dysfunction and axonal degeneration [20]. Loss of SIGMAR1 was related to impaired mitochondria–ER interactions and axonal degeneration, while the inhibition of mitochondrial fission was implicated in axonal degeneration through affected intracellular Ca2+ signaling, activation of ER stress and impaired mitochondrial dynamics [18]. Although the size of mitochondria–ER juxtapositions was similar in degenerating and non-degenerating axons, it requires further analyses to address if the functions of MAM are changed upon degenerating the demyelinated axons. Future studies are necessary to address if interventions that enhance mitochondria–ER interactions facilitate functional maintenance and improve the survival of demyelinated axons, and thus could be useful as therapeutic strategies in myelin disorders.
References
Nave KA (2010) Myelination and support of axonal integrity by glia. Nature 468:244–252
Trapp BD, Nave KA (2008) Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 31:247–269
Lassmann H, Bruck W, Lucchinetti C (2001) Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med 7:115–121
Waxman SG (2006) Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci 7:932–941
Kiryu-Seo S, Ohno N, Kidd GJ, Komuro H, Trapp BD (2010) Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J Neurosci 30:6658–6666
Mahad DH, Trapp BD, Lassmann H (2015) Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 14:183–193
Ohno N, Chiang H, Mahad DJ, Kidd GJ, Liu L, Ransohoff RM, Sheng ZH, Komuro H, Trapp BD (2014) Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc Natl Acad Sci USA 111:9953–9958
Zambonin JL, Zhao C, Ohno N, Campbell GR, Engeham S, Ziabreva I, Schwarz N, Lee SE, Frischer JM, Turnbull DM, Trapp BD, Lassmann H, Franklin RJ, Mahad DJ (2011) Increased mitochondrial content in remyelinated axons: implications for multiple sclerosis. Brain 134:1901–1913
Hoppins S, Lackner L, Nunnari J (2007) The machines that divide and fuse mitochondria. Annu Rev Biochem 76:751–780
Friedman JR, Nunnari J (2014) Mitochondrial form and function. Nature 505:335–343
Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009) MAM: more than just a housekeeper. Trends Cell Biol 19:81–88
Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14:759–774
Rizzuto R, De Stefani D, Raffaello A, Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13:566–578
Theurey P, Rieusset J (2017) Mitochondria-associated membranes response to nutrient availability and role in metabolic diseases. Trends Endocrinol Metab 28:32–45
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610
Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P (2009) An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325:477–481
Rowland AA, Voeltz GK (2012) Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13:607–625
Bernard-Marissal N, Medard JJ, Azzedine H, Chrast R (2015) Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138:875–890
van Vliet AR, Verfaillie T, Agostinis P (2014) New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta 1843:2253–2262
Yin X, Kidd GJ, Ohno N, Perkins GA, Ellisman MH, Bastian C, Brunet S, Baltan S, Trapp BD (2016) Proteolipid protein-deficient myelin promotes axonal mitochondrial dysfunction via altered metabolic coupling. J Cell Biol 215:531–542
Kagawa T, Ikenaka K, Inoue Y, Kuriyama S, Tsujii T, Nakao J, Nakajima K, Aruga J, Okano H, Mikoshiba K (1994) Glial cell degeneration and hypomyelination caused by overexpression of myelin proteolipid protein gene. Neuron 13:427–442
Thai TQ, Nguyen HB, Saitoh S, Wu B, Saitoh Y, Shimo S, Elewa YH, Ichii O, Kon Y, Takaki T, Joh K, Ohno N (2016) Rapid specimen preparation to improve the throughput of electron microscopic volume imaging for three-dimensional analyses of subcellular ultrastructures with serial block-face scanning electron microscopy. Med Mol Morphol 49:154–162
Cardona A, Saalfeld S, Schindelin J, Arganda-Carreras I, Preibisch S, Longair M, Tomancak P, Hartenstein V, Douglas RJ (2012) TrakEM2 software for neural circuit reconstruction. PLoS One 7:e38011
Ohno N, Kidd GJ, Mahad D, Kiryu-Seo S, Avishai A, Komuro H, Trapp BD (2011) Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J Neurosci 31:7249–7258
Nguyen HB, Sui Y, Thai TQ, Ikenaka K, Oda T, Ohno N (2018) Decreased number and increased volume with mitochondrial enlargement of cerebellar synaptic terminals in a mouse model of chronic demyelination. Med Mol Morphol 51:208–216
Marchi S, Patergnani S, Pinton P (2014) The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta 1837:461–469
Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2015) Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci USA 112:E2174–E2181
Leal NS, Schreiner B, Pinho CM, Filadi R, Wiehager B, Karlstrom H, Pizzo P, Ankarcrona M (2016) Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid beta-peptide production. J Cell Mol Med 20:1686–1695
de Brito OM, Scorrano L (2010) An intimate liaison: spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J 29:2715–2723
Inoue K (2005) PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics 6:1–16
Willard HF, Riordan JR (1985) Assignment of the gene for myelin proteolipid protein to the X chromosome: implications for X-linked myelin disorders. Science 230:940–942
Readhead C, Schneider A, Griffiths I, Nave K-A (1994) Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron 12:583–595
Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, Lassmann H, Turnbull DM (2009) Mitochondrial changes within axons in multiple sclerosis. Brain 132:1161–1174
Mutsaers SE, Carroll WM (1998) Focal accumulation of intra-axonal mitochondria in demyelination of the cat optic nerve. Acta Neuropathol 96:139–143
Sathornsumetee S, McGavern DB, Ure DR, Rodriguez M (2000) Quantitative ultrastructural analysis of a single spinal cord demyelinated lesion predicts total lesion load, axonal loss, and neurological dysfunction in a murine model of multiple sclerosis. Am J Pathol 157:1365–1376
Witte ME, Bo L, Rodenburg RJ, Belien JA, Musters R, Hazes T, Wintjes LT, Smeitink JA, Geurts JJ, De Vries HE, van der Valk P, van Horssen J (2009) Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol 219:193–204
Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernandez-Alvarez MI, Zorzano A, De Stefani D, Dorn GW, Scorrano L (2016) Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci USA 113:11249–11254
Szymanski J, Janikiewicz J, Michalska B, Patalas-Krawczyk P, Perrone M, Ziolkowski W, Duszynski J, Pinton P, Dobrzyn A, Wieckowski MR (2017) Interaction of mitochondria with the endoplasmic reticulum and plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int J Mol Sci 18:1576
Campbell G, Mahad DJ (2018) Mitochondrial dysfunction and axon degeneration in progressive multiple sclerosis. FEBS Lett 592:1113–1121
Acknowledgements
This work is partly supported by JSPS KAKENHI Grant Number 16K18977 (to N.O.), Cooperative Research Program of “Network Joint Research Center for Materials and Devices”, Research Grant from National Center of Neurology and Psychiatry (No. 30-5 to N.O.) and Cooperative Study Programs of National Institute for Physiological Sciences (to N.O.). We would like to thank Setsuro Fujii Memorial, Osaka Foundation for Promotion of Fundamental Medical Research, for providing the support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Thai, T.Q., Nguyen, H.B., Sui, Y. et al. Interactions between mitochondria and endoplasmic reticulum in demyelinated axons. Med Mol Morphol 52, 135–146 (2019). https://doi.org/10.1007/s00795-018-0212-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00795-018-0212-0